Introducción
El sarcoma de Ewing (SE) es el segundo tumor óseo maligno más común en niños y adolescentes de huesos largos, la tasa de supervivencia alcanza hasta el 70% de los casos, sin embargo, rara vez su presentación se da en sitios distintos a huesos largos. El sarcoma de Ewing puede surgir en huesos como la pelvis, el fémur, la tibia y las costillas. Además, se puede presentar en tejidos blandos como en la pared torácica, el músculo glúteo, cavidades pleurales y músculos cervicales, en menor proporción1.
La presentación clínica del sarcoma de Ewing difiere entre los grupos de edad según factores relacionados que incluyen el sexo, el estadio y el tamaño del tumor. A continuación, se describe un caso de sarcoma de Ewing metastásico que mostró una rápida progresión de la enfermedad sistémica con metástasis esqueléticas generalizadas sin evidencia de compromiso pulmonar. Creemos que esta progresión atípica y rápida de la enfermedad (luego de un mes del diagnóstico) no se ha informado previamente en la literatura ortopédica, además del sitio inicial de la lesión en región frontoparietal derecha1.
Un enfoque interdisciplinario ha dado como resultado una mejora significativa en el pronóstico de los pacientes con estos tumores. A pesar del tratamiento agresivo, el 20-40% de los pacientes con enfermedad localizada y casi el 80% de los pacientes con enfermedad metastásica al momento de la presentación perecen a la enfermedad. En la actualidad los avances en la comprensión de la biología molecular de estos tumores dan como resultado el desarrollo de nuevos enfoques de tratamiento. El objetivo de este artículo es resaltar la presentación clínica atípica como parte del diagnóstico y los métodos de tratamiento existentes. Un enfoque interdisciplinario ha dado como resultado una mejora significativa en el pronóstico de los pacientes con estos tumores2,3.
Caso clínico
Paciente de 15 años ingresa remitida a nuestra institución, con cuadro clínico de tres meses de evolución de cefalea, edema orbital derecho y tumefacción temporoparietal derecha, por lo cual consultan a hospital local de su comunidad en varias ocasiones, dando manejo sintomático con analgesia, sin mejoría, por lo cual realizan ecografía de tejidos blandos en julio del 2022 con reporte de lesión con características benignas posiblemente un lipoma.
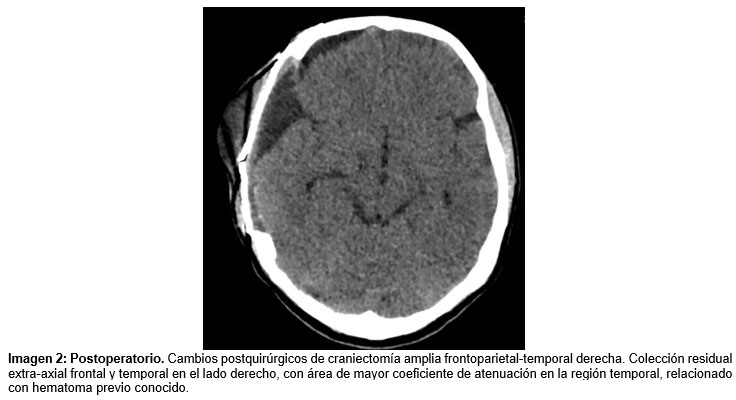
Posteriormente fue manejada con AINES (antiinflmatorios no esteroideos), pero con aumento en tamaño de la lesión y dolor incrementando con el tiempo hasta que decidió consultar nuevamente, en esta ocasión se realiza tomografía axial computarizada (TAC) de cráneo el 03/09/2022 con reporte de lesión extra axial heterogénea que compromete la región parietal temporal y frontal derechas de 82*28*62 mm con patrón permeativo y alteración de los tejidos blandos adyacentes con realce en forma heterogénea con medio de contraste. Debido a este reporte le indican a madre que requiere otro nivel de atención debido a la complejidad de la lesión y la remiten a nuestra institución. Se realiza imagen con TAC antes del manejo quirúrgico (Imagen 1).
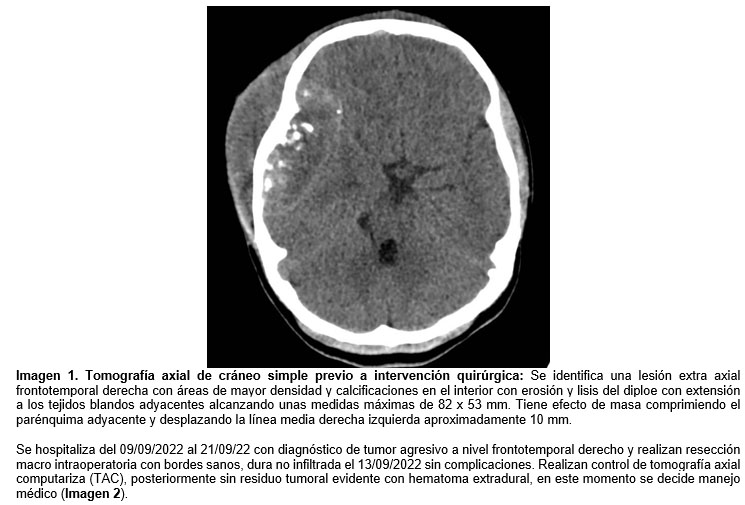
Se recibe reporte de patología el 23/09/2022 donde describen lesión tumoral indiferenciada con informe de inmunohistoquímica que confirma tumor de Ewing por lo que neurocirugía redirecciona para hospitalización y manejo conjunto por oncología pediátrica.
El reporte de patología del 13/09/2022 de inmunohistoquímica: la población de células neoplásicas es positivas para vimentina, BCL2, CD99 en tinción difusa y fuerte de membrana y FLI1. Se conserva la expresión del INI1 y el KI67 es positivo en aproximadamente el 50%. El resto de los marcadores son negativos. Por morfología e inmunohistoquímica hay elementos para confirmar el diagnóstico de un sarcoma de Ewing.
Se confirma sarcoma de Ewing frontotemporal intervenido quirúrgicamente sin complicaciones y con resección completa por imágenes postquirúrgicas. Se solicitaron imágenes de extensión de tomografía de abdomen y tórax contrastados, PET - CT y gammagrafía ósea. En las cuales se evidencia compromiso metastásico, a nivel del cráneo cambios posquirúrgicos sin evidencia de recaída-residuo tumoral hipermetabólico. Con adenopatía del lado izquierdo del cuello con incremento metabólico. Lesiones pancreáticas hipermetabólicas con características sospechosas de compromiso metastásico dada su multifocalidad. Compromiso óseo a nivel de pelvis y fémur izquierdo. Lesión subpleural inflamatoria cicatricial en pulmón izquierdo. Actualmente en manejo con protocolo de quimioterapia, en seguimiento clínico.
Discusión
El sarcoma de Ewing es el segundo tumor óseo primario más común que se observa en niños y adolescentes, fue descrito por James Ewing en 1921 como un endotelioma óseo difuso. Es uno de los diagnósticos diferenciales de los tumores pediátricos de células azules pequeñas y redondas. Esta no es una condición única, sino un grupo de trastornos estrechamente relacionados morfológica y clínicamente con una biología molecular similar (expresión de oncoproteínas quiméricas específicas de tumores a través de translocaciones cromosómicas equilibradas que involucran el gen EWS), a menudo denominado familia de tumores de Ewing. Esto incluye el sarcoma óseo de Ewing, el sarcoma de Ewing extraóseo, el tumor de Askin y el tumor neuroectodérmico periférico. Estas son neoplasias agresivas con casi el 25% de los pacientes que tienen metástasis clínicamente evidentes en la presentación. El sarcoma de Ewing se ha considerado como una enfermedad sistémica que requiere tratamiento tanto local como sistémico2,4.
La localización intracraneal primaria es extremadamente rara, comprende aproximadamente el 1-4% de los casos. Se origina más comúnmente en la duramadre con una gran lesión que causa déficit neurológico. Hasta donde se encontró, hay solo hay cuatro casos de SE en la literatura con localización en hueso mastoideo y ninguno de estos pacientes en menores de cinco años. Dos de estos se originaron en el hueso temporal y ninguno de ellos en menores5,6. Nuestro caso llama la atención la ubicación a nivel de región temporoparietal derecha, ya que no es la ubicación común de esta patología, incluso inicialmente se manejó y se diagnosticó como una lesión benigna, siendo esta más común por las características clínicas iniciales.
Después de los estudios de diagnóstico por imágenes, se debe obtener una biopsia para confirmar el diagnóstico histopatológico de SE; como en nuestro caso, este se caracteriza por ser tumores indiferenciado de células redondas azules con citoplasma escaso y núcleo hipercromático. Al igual que los estudios inmunohistoquímicos como marcadores, vimentina, HBA-71 y microglobulina B2 su positividad ayuda a distinguir el SE de otros tumores5, lo cual se realizó en nuestra paciente dando positividad para vimentina, BCL2, CD99.
El tratamiento estándar para SE incluye quimioterapia sistémica, cirugía y/o radioterapia (RT) como se realizó en nuestro caso. En lactantes no hay recomendaciones estándar para el tratamiento. En pacientes tan jóvenes, el objetivo del tratamiento es obtener el máximo posibilidad de curación con un riesgo mínimo de toxicidad. El SE es una enfermedad muy rara en el grupo etario distinto a adolescentes y se desarrolla en varias partes del cuerpo; sin embargo, ocurre más frecuentemente en el esqueleto axial 5, a diferencia de nuestro caso, cuya presentación debut se dió como una lesión en región frontoparietal derecha, por lo cual inicialmente no se manejó como una lesión maligna.
Actualmente, el tratamiento estándar consiste en quimioterapia sistémica y terapia local la primera línea consiste en quimioterapia con múltiples agentes que incluye vincristina, doxorrubicina y ciclofosfamida, alternando con ifosfamida y etopósido administrado durante aproximadamente 12 semanas antes de la terapia local definitiva y varios meses después. Es muy importante el tratamiento local que consiste en cirugía con una máxima resección o RT, o una combinación de ambos para lograr un control local óptimo. Cuando sea posible, la resección quirúrgica máxima debe ser realizada, sin embargo, la proximidad de estructuras vitales impide la cirugía completa implicando mal pronóstico. El SE es un tumor radiosensible por lo que la RT se puede utilizar como único tratamiento, en los tumores que no pueden ser resecados7.
En la literatura, los pacientes tratados solamente con RT tuvieron resultados significativamente peores. Bacci et al. Encontraron que la tasa de fracaso local era 19%, 9% y 11% en pacientes tratados con RT sola, cirugía sola y cirugía y RT, respectivamente. Sin embargo, muchos pacientes tenían tumores inoperables en condiciones desfavorables debido a la ubicación anatómica (pelvis, columna vertebral, intracraneal, etc.) lo que puede haber sesgado los resultados, por lo cual a nivel general, sigue siendo la terapia combinada la que ha demostrado mejores resultados8. En nuestro caso en primer lugar el manejo quirúrgico y luego el inicio de quimioterapia fue la pauta establecida según protocolos actuales.
Alrededor del 25% de los pacientes con SE presenta enfermedad metastásica en el momento del diagnóstico. El sitio común en la mayoría de las metástasis es el pulmón, seguido por hueso. Aunque el SE de región intracraneal se relaciona con menor frecuencia con metástasis al momento del diagnóstico9. Nuestro caso se caracteriza por una evolución rápida y con metástasis confirmadas pocos meses después del momento del diagnóstico.
Los protocolos de tratamiento actuales también incluyen enfoque de tratamiento curativo para los pacientes con enfermedad metastásica. RT de pulmón completo después de una respuesta completa a la convencional. La quimioterapia es un tratamiento estándar para pacientes con enfermedad metastásica limitada a nivel pulmonar5,10.
En conclusión, el sarcoma de Ewing y el tumor neuroectodérmico primitivo son raros, se agrupan bajo el espectro de un conjunto de tumores. Estos son altamente malignos involucran los huesos y comúnmente ocurren en niños, en el grupo de adolescentes. El sarcoma de Ewing del hueso del cráneo representa solo el 1 % de todos los sarcomas de Ewing. Uno de los motivos por los cuales se expone nuestro caso.
Por lo tanto, una combinación de apariencia histopatológica, hallazgos radiográficos y correlación clínica es importante para determinar el diagnóstico correcto, establecer el régimen de tratamiento adecuado y mejorar la supervivencia del paciente. Un comportamiento de rápida progresión, agresivo, se ha relacionado con mutaciones específicas, cada día se relaciona más el genotipo y fenotipo en las enfermedades y esta patología no es la excepción.
Bibliografía
- Al-Marshad AY, AlMazrua IS, AlMulla F, Alaboud M, Alrifaie O, Shaheen M, et al. Unusual presentation of Ewing sarcoma; a case report and literature review. Int J Surg Case Rep. octubre de 2022;99:107479.
- Thacker MM, Temple HT, Scully SP. Current treatment for Ewing’s sarcoma. Expert Rev Anticancer Ther. abril de 2005;5(2):319-31.
- Rachapudi S, Srinivasan A, Gorman B, Wilhelm AB, Eyzaguirre E, Orihuela E. Ewing’s sarcoma of the male external genitalia: a case report and review of the literature. BMC Urol. 8 de agosto de 2022;22(1):121.
- Machado I, Noguera R, Mateos EA, Calabuig-Fariñas S, López FIA, Martínez A, et al. The many faces of atypical Ewing’s sarcoma. A true entity mimicking sarcomas, carcinomas and lymphomas. Virchows Arch. marzo de 2011;458(3):281-90.
- Esen ÇSB, Gültekin M, Aydın GB, Akyüz C, Oğuz KK, Orhan D, et al. Ewing sarcoma in an infant and review of the literature. Turk J Pediatr. 2019;61(5):760.
- Müngen E, Kurucu N, Kutluk T, Oğuz KK, Söylemezoğlu F, Yalçın B. Primary spinal multifocal intradural-extramedullary ewing sarcoma in children: presentation of a case and review of the literature. Turk J Pediatr. 2021;63(6):1084.
- Eaton BR, Claude L, Indelicato DJ, Vatner R, Yeh B, Schwarz R, et al. Ewing sarcoma. Pediatr Blood Cancer [Internet]. mayo de 2021 [citado 12 de noviembre de 2022];68(S2). Disponible en: https://onlinelibrary.wiley.com/doi/10.1002/pbc.28355
- Gotsmy W, Neumayer B, Kraus T, Zellinger B, Neureiter D, Monticelli F, et al. Fatal course of undetected Ewing-like sarcoma in a 9-year-old boy with uncharacteristic clinical presentation. Forensic Sci Med Pathol. septiembre de 2022;18(3):231-9.
- Jo VY. EWSR1 fusions: Ewing sarcoma and beyond. Cancer Cytopathol. abril de 2020;128(4):229-31.
- Saifuddin MSAH, Ng CY, Abdullah MS. Skull Base Primary Ewing Sarcoma: A Radiological Experience of a Rare Disease in an Atypical Location. Am J Case Rep [Internet]. 4 de mayo de 2021 [citado 27 de octubre de 2022];22. Disponible en: https://www.amjcaserep.com/abstract/index/idArt/930384
|
