Introducción
El término Enfermedades Inflamatorias Intestinales (EII) incluye a la Enfermedad de Crohn (EC), Colitis Ulcerosa (CU) y Colitis indeterminada (CI) (1), que son una importante causa de patología gastrointestinal en niños y adolescentes. La incidencia de éstas se encuentra en aumento en la edad pediátrica (2), manteniéndose en los países en que históricamente se ha observado una alta incidencia y aumentando en los países en vías de desarrollo, como el nuestro (3). En las últimas décadas, el comportamiento epidemiológico de ambas enfermedades ha sido diferente, la CU se ha mantenido estable, mientras que la EC ha aumentado en la población pediátrica (4).
Diferencias de incidencia a través de la edad, el sexo y regiones geográficas, sugieren la existencia de factores ambientales que modifican significativamente la expresión de las EII (3).
Estas patologías se caracterizan por ser entidades clínicas, que afectan al sistema gastrointestinal, compartiendo entre sí una evolución crónica con períodos de exacerbaciones y remisiones asociados a una activación desbalanceada del sistema inmune de la mucosa intestinal en individuos genéticamente predispuestos (5).
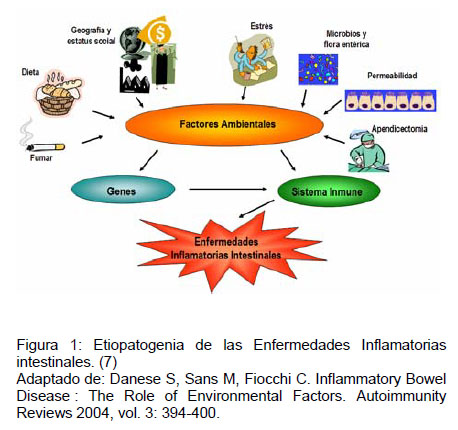
La etiopatogenia de EII no esta completamente dilucidada por lo que actualmente se considera que es de etiología multifactorial (6). Se han planteado diversos mecanismos y determinantes que podrían estar involucrados en la génesis de estas patologías, que incluyen: factores ambientales, familiares, genéticos y del sistema inmune. Con respecto a los factores ambientales implicados se postula que tanto la dieta, el tabaco, uso de ciertos medicamentos, la apendicectomía, factores psicológicos como el estrés y algunos estilos de vida, tendrían alguna implicancia en la susceptibilidad a desarrollar alguna forma de EII (6,7,8). La agregación familiar es el mayor factor de riesgo independiente para desarrollar EII (9) Desde el punto de vista de la genética, las EII se consideran enfermedades poligénicas, atribuyéndose algún rol a múltiples genes (6). Alteraciones de la inmunidad incluyen: pérdida la tolerancia a la flora intestinal comensal y alteraciones de la respuesta inmunológica tanto innata como adquirida (10).
En esta revisión se pretende exponer el comportamiento epidemiológico de las EII, con énfasis en la población pediátrica, y también revisar algunos de los mecanismos que podrían estar involucrados en la etiopatogenia de estas patologías.
Incidencia y Prevalencia
Históricamente los países que tienen las mayores tasas de incidencia y prevalencia de EII son Estados Unidos, el Reino Unido y países de norte de Europa. En Estados Unidos se observa una incidencia que fluctúa entre los 2,2-14,3 casos por 100.000 habitantes para CU y entre 3,1-14,6 por 100.000 habitantes para EC. La prevalencia de las EII en el misma región se encuentra entre 37-246 por 100.000 habitantes para CU y entre 26-199 casos por 100.000 habitantes para la EC (3).
Clásicamente se describían lugares en el mundo (ej: América Latina) en que las EII eran una entidad clínica poco frecuente, sin embargo, las tasas de incidencia y prevalencia han tenido un franco aumento, en las últimas décadas (11). Se presume que este fenómeno se debe a cambios ambientales como son la dieta y los estilos de vida, entre otros (3,7). En las últimas décadas, el comportamiento epidemiológico de ambas enfermedades ha sido diferente, la CU se ha mantenido estable, mientras que la EC ha aumentado en la población pediátrica (4,12).
Características Demográficas
Género: En pacientes pediátricos no existen diferencias significativas entre ambos sexos en CU. Mientras que en EC existe un ligero predominio del sexo femenino (12).
Edad: La edad del diagnóstico de las EII, tiene un comportamiento bimodal, con el primer peak en la tercera década de la vida y el segundo entre la quinta y séptima década (12,13) aunque se puede presentar a cualquier edad (3). El diagnóstico de EII se efectúa principalmente en adultos jóvenes, sin embargo existe un 15-25% de los casos que debutan durante la edad pediátrica (12). Un estudio realizado entre los años 2000 y 2001, en población pediátrica de Wisconsin (14), encontró que el promedio de edad al momento del diagnóstico de
EII era de 12.5 años. En esa misma serie se objetivó que la edad promedio al momento del diagnostico en EC fue de 13.5 (±4.5) años y de 11.8 (±4.4) años para CU (14).
Etiopatogenia
El origen de las EII es el resultado de la interacción de factores ambientales, familiares, genéticos y de la inmunidad (Figura 1). A continuación se expondrá como interviene cada uno de estos factores en su etiopatogenia.
Factores Ambientales
El aumento de la incidencia de las EII a nivel mundial, en un período corto de tiempo, ha motivado el estudio sistemático de los factores ambientales y de su responsabilidad en este fenómeno, ya que esto no puede ser explicado del todo por variaciones genéticas (7,8). Entre los factores involucrados se mencionan:
Tabaco : El tabaco tiene efectos opuestos en CU y EC (8). En CU el tabaco se comporta como factor protector tanto en el desarrollo de la enfermedad como en la prevención de exacerbaciones (6). Se ha demostrado además, que el cese del hábito se relaciona con un incremento del riesgo de desarrollar CU (7). Por el contrario, el fumar complica la evolución de pacientes con EC, aumentando las exacerbaciones ya que promueve la formación de fístulas, estenosis e incrementa el uso de corticoesteroides (6,7). A su vez, adelanta la necesidad del tratamiento quirúrgico en la evolución de la enfermedad. El cese del hábito ha demostrado mejorar la evolución clínica de la enfermedad (7). El efecto paradójico del hábito tabáquico apoya el argumento de que existirían distintos mecanismos involucrados en la etiopatogenia de estas dos enfermedades, aunque aun no se ha llegado a una conclusión satisfactoria al respecto (8).
Dieta : No se ha podido establecer con certeza cómo afecta la dieta en la etiopatogenia de EII. Existen reportes de la existencia de una relación directa de una dieta rica en azúcares refinados y grasas poli-insaturadas con la enfermedad. Pero la evidencia con respecto a este factor es débil aún, por las dificultades de metodología que implica estudiar este factor de riesgo (3,7,8).
Fármacos : Entre los fármacos que podrían tener relación con las EII, están los anticonceptivos orales (ACO) y los anti-inflamatorios no esteroidales (AINES). Con respecto a los primeros existen reportes de que las pacientes usuarias de ACO, tienen mayor riesgo de desarrollar EII. Sin embargo no esta claro cuál podría ser la relación directa con el desarrollo de EII (3,7). Diferente es el caso de AINES, puesto que existe una clara asociación como factor de riesgo. En esta línea, pacientes con remisión clínica de EII recaen en mayor medida luego del uso de AINES. Existen estudios en ratones knock-out para el gen de IL-10 que desarrollan espontáneamente un tipo de colitis similar a CU, en los que el curso de la patología en presencia de AINES es más agresivo (7).
Factores geográficos y socio-económicos : Al respecto, estudios epidemiológicos describen una incidencia mayor, aunque estable, en Estados Unidos, Inglaterra y países de Europa oriental (3,4). Con una tendencia al alza en regiones de Asia y el sur de Europa (CU). Se ha relacionado a grupos de mayores ingresos económicos con una mayor prevalencia de EII, así como también a personas que realizan trabajos livianos durante largos períodos de tiempo, no así en las personas con trabajos activos (7).
Stress : Se le ha atribuido un rol en el comportamiento clínico y evolución de la enfermedad más que en la génesis de EII. Evidenciándose en clínica un incremento de la tasa de exacerbaciones en periodos de stress sostenido en el tiempo. Observaciones en animales apoyan estas aseveraciones (7).
Factores Microbiológicos : Existen reportes que ciertas infecciones atribuyen a un rol en la etiopatogenia, como las provocadas por: L. monocytogenes, Chlamydia Trachomatis, E. Coli, Saccharomyces cerevisae (10). Sin embargo, la bacteria Mycobacterium Paratuberculosis representa un caso especial, puesto que genera una infección intestinal en ovejas, cuyas características anatomopatológicas se asemejan a las lesiones propias de la EC , motivando el estudio de su implicancia en el desarrollo de la enfermedad (3,10). Se ha propuesto la relación entre infección viral y EII, puesto que se han aislado partículas virales en granulomas de EC. Los agentes etiológicos virales aislados forman parte de la familia Paramixoviridae, como el virus Sarampión. Se han reportado asociaciones de mayor incidencia de EII en pacientes que recibieron la vacuna a virus vivo contra el Sarampión (8). Sin embargo, esta asociación ha caído en descrédito por nuevos estudios que han obtenidos resultados contradictorios (3,7). Además, el aumento de la incidencia de EII durante los últimos años contrasta con la disminución concomitante de estas infecciones (6,7).
Apendicectomía : Existen estudios que revelan que la apendicectomía sería un factor protector del desarrollo en CU. Evidencia en animales es concordante con esta observación aunque los mecanismos involucrados no están completamente dilucidados. En contraparte existen estudios que la asocian como factor de riesgo para el desarrollo de EC (7).
Factores Familiares
La hipótesis de que un determinado genotipo seria el responsable de la susceptibilidad a desarrollar una EII tiene su raíz en estudios de familias que demuestran que la presencia de al menos un familiar con EII como antecedente, es factor de riesgo para el desarrollo de la enfermedad (6). Un estudio (14) en pacientes pediátricos reveló que un 11% los niños diagnosticados recientemente tenia al menos un pariente en primer o segundo grado afectado con EII. La presencia en la historia clínica de antecedentes familiares, cobra mayor importancia en niños y adolescentes, ya que además se ha demostrado que el tener un familiar afectado se relaciona con un inicio precoz de la enfermedad (15). En cuanto al grado de parentesco, el antecedente de un familiar en primer grado afectado con la misma EII, está presente entre un 2,2-16% en pacientes con EC, y entre un 5,7-15% en CU (6). Resultados de un estudio de cohorte de pacientes pediátricos (n=1000) refieren que un 30,7% de los niños afectados, tienen parientes en cualquier grado con historia de EII mientras que el 16% tienen algún familiar en primer grado (16).
Estudios de concordancia en gemelos sugieren una influencia genética en la patogenia de EII, siendo más notorio en EC con un 37,3% de concordancia en gemelos monocigóticos mientras que para CU alcanza un 10%. En el caso de gemelos dicigóticos se alcanzan valores de 7% para EC y 3% para CU (6).
Factores Genéticos
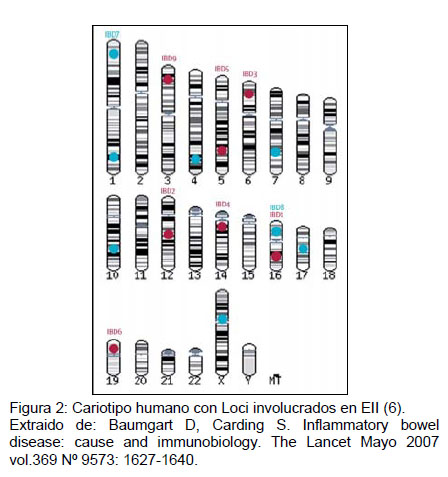
En la literatura las EII, se consideran enfermedades poligénicas. Se han identificado regiones en 12 cromosomas distintos que tendrían implicancia en la patogenia de EC y CU (Figura 2) (6). Sin embargo, dentro de las áreas del genoma relacionadas a EII aparecen algunos genes a los que se les ha atribuido un grado mayor de responsabilidad en cuanto al desarrollo de la enfermedad.
CARD 15/NOD2 : Es el caso de mutaciones en el gen CARD 15 que codifica a la proteína NOD 2 ( nucleotide-binding oligomerization domain 2 ) ubicado en el cromosoma 16. A esta proteína se le ha conferido responsabilidad en la génesis de EC (5). La molécula NOD 2 es una proteína intracelular que permite detectar la presencia de elementos bacterianos y activar la cascada de respuesta de la inmunidad innata. Mutaciones en NOD-2 afectan la capacidad de esta proteína de activar adecuadamente la expresión de NF?B (5). La adecuada expresión de NF?B es necesaria para mantener una adecuada homeostasis intestinal, activando la respuesta inmune frente a microorganismos patógenos. Se postula que el déficit de expresión de NF?B determinaría una respuesta inflamatoria inadecuada y descontrolada, desencadenando la expresión de citoquinas y otras moléculas pro-inflamatorias que gatillarían la enfermedad (5,6).
IBD-5 : Existe otro gen relacionado con EII, denominado IBD-5 ( Inflammatory bowel Disease -5 ). Ubicado en el cromosoma 5 que codifica dos proteínas transmembrana OCTN1 y OCTN 2 (Organic Cationic Transporter ) cuya función es la de traslocar carnitina y otras sustancias al interior de la célula (6). Un estudio demostró que mutaciones en estas proteínas fueron encontradas en el 53% de pacientes con EC versus un 23% en personas libres de enfermedad. Se postula que una disfunción de esta molécula aumentaría el transporte a interior de la célula de componentes inmunogénicos, provenientes de bacterias patógenas, resultando en una exacerbación de la respuesta inmunológica (17).
Genotipo/Fenotipo : Con respecto a la diversidad de presentación clínica que pueden caracterizar a las EII, existen múltiples genes que se han asociado a ellas, ya sea en cuanto a su gravedad, pronóstico o forma de presentación. En esta categoría cabe mencionar que mutaciones en NOD-2 se asocian a exclusivamente a EC en pacientes de raza blanca y a la estenosis de intestino (6). El halotipo DRB *0103 ha sido relacionado con un curso particularmente agresivo de CU y con la necesidad de cirugía en enfermedad con compromiso de colon en EC (6,8). Algunas de las manifestaciones extraintestinales de las EII, como la espondilitis anquilosante, se han asociado con mutaciones de las proteínas codificadas en la región HLA- B27 (18). Mientras que la Uveítis se ha relacionado con HLAB44 o HLA-DRB*0103 (6).
Factores Inmunológicos
Los desbalances inmunológicos observados en EII corresponden a alteraciones tanto en la inmunidad innata como en la adquirida (5). En este sentido, se le ha atribuido a la interacción del sistema inmunológico intestinal con la flora comensal, un rol central en la patogénesis de la EII.
La pérdida de la tolerancia del sistema inmunológico intestinal frente a la flora comensal seria un factor clave en la etiopatogenia de la EII (19).
En condiciones normales, el sistema inmunológico del intestino esta en permanente contacto con más de 500 diferentes especies de Bacterias (6), y es capaz de discriminar entre la microbiota normal intestinal y bacterias patógenas, manteniendo una tolerancia inmunológica para las primeras y montando una respuesta totalmente diferente con las segundas, con el fin de erradicarlas del organismo (10).
La hipótesis que involucra a la flora comensal, con la patogenia de las EII se basa en observaciones en ratones manipulados con ingeniería genética que desarrollan EII en presencia de bacterias de la flora comensal, pero que no lo hacen en un ambiente estéril (5,11,18). En la misma línea existe evidencia en animales, de remisión de la enfermedad con el uso de antimicrobianos de amplio espectro (11).
A la luz de estos hallazgos se plantea que existiría una pérdida de la tolerancia frente a bacterias comensales la que jugaría un rol central en la etiopatogenia de EII (5,6,11,18). En apoyo de esta hipótesis existe evidencia de pacientes con EII que poseen una gran concentración de anticuerpos en la mucosa intestinal, contra antígenos de microbiota comensal (18). Algunos estudios se inclinan ante la posibilidad de que la infección por ciertas bacterias patógenas estaría implicada en la activación de la inmunidad celular y humoral desencadenado la enfermedad, como algunos serotipos de E.coli, Mycobacterium, Listeria y Yersinia (20).
El mecanismo propuesto para esta hipótesis, es el montaje de una respuesta inmune como consecuencia de la presentación de antígenos del lumen intestinal (de la flora comensal o de otra fuente) a linfocitos T-helper, con su posterior activación y producción de citoquinas (10). Las citoquinas liberadas desencadenarían la activación de linfocitos T CD8+ o CD4+ determinando el estado inflamatorio característico de la EII (5,10).
Las condiciones que determinan la susceptibilidad a desarrollar esta respuesta alterada ante antígenos presentes en el lumen intestinal se describen a continuación:
Alteración de Inmunidad Innata:
Alteraciones o debilidad de la barrera epitelial: Las células epiteliales intestinales están recubiertas por mucus, constituyendo la primera línea de defensa contra agresiones del medio externo. Se ha reportado que los pacientes con EII poseen una deficiencia en la producción de mucus (6,8). Esta condición se ha relacionado más a CU que a EC (8). Algunas alteraciones en la permeabilidad de la barrera intestinal que involucran a las uniones estrechas (Tight-junction) , encargadas de mantener la barrera mucosa intestinal han sido descritas (6). Esta debilidad de la barrera epitelial tendría como resultado una estimulación continua del sistema inmunológico intestinal, gatillando el proceso inflamatorio crónico (5,6).
Toll Like Receptors (TLR's): Existe evidencia que pacientes con EII poseen un patrón de expresión de TLRs alterado (6). Dichos receptores, reconocen elementos microbianos o moléculas asociadas a ellos como Lipopolisacáridos (LPS) peptidoglicanos, ácido lipoteicoico entre otros elementos comunes a muchas bacterias y que permiten el inicio de la cascada de la respuesta innata y posteriormente de la adaptativa. En ausencia de patógenos, los TLR participan en la mantención de la tolerancia de la microbiota comensal y la mantención de la barrera epitelial (6). En personas sanas los sub-tipos TLR5 y TLR3 son expresadas en la membrana basolateral de las células epiteliales, mientras que TLR2 y TLR4 son escasamente detectables. Esto contrasta con la sobrexpresión de TLR4 en pacientes con EII, y la baja expresión de TLR3 en EC, no así en CU. Estas alteraciones podrían determinar una falla en la respuesta normal frente a bacterias patógenas impidiendo su eliminación y propiciando un estado de inflamación crónica (6).
Alteraciones de la inmunidad adaptativa:
El sistema inmune normal, está en permanente contacto con innumerables antígenos de alimentos y microbiota comensal, sin provocar una respuesta inflamatoria, estableciendo un estado de anergia frente a esos desafíos. Esta situación, permite postular que existe una alteración en el funcionamiento de células implicadas en el reconocimiento, presentación y regulación de la respuesta inmune adaptativa, en las EII (5). Existen estudios en modelos animales en que las células dendríticas, principales agentes presentadores de antígenos, reconocen en forma anormal antígenos de bacterias comensales e inducen una respuesta inmunológica, que incluye la liberación de citoquinas propias de respuesta de tipo Th1 (Il-2 Il-1 Il-6 IL-12,IL-18, TNF-a e INF-?) (6,10). Esta hipótesis está sustentada en la evidencia de que en pacientes con EII existen niveles aumentados de estas citoquinas en tejido intestinal así como también en sangre periférica (8,10). En el caso de CU, se atribuye a una respuesta mixta Th1 y Th-2 caracterizada por la secreción de IL-4, IL-5 e IL-10 (10,11).
Entre las funciones que desempeña el TNF-a en la modulación de la respuesta inmunológica destacan: la amplificación de la respuesta inmune, mediante la estimulación de la secreción de IL-6 e IL-1B, expresión de moléculas de adhesión, proliferación de fibroblastos y como iniciador de respuestas citotóxicas e inductor de apoptosis. También es el responsable de inducir la secreción de INF-?. La gran cantidad de funciones que desempeña en la cascada inmunitaria lo ha identificado como un blanco terapéutico, mediante la utilización de anticuerpos Anti TNF-a (10). Estudios revelan una expresión elevada de TNF-a en pacientes con EII (5,21) y relacionan su incremento con la actividad clínica de la enfermedad. En modelos de ratones knock-out para TNF-a, se ha logrado impedir el desarrollo de colitis y en modelos murinos en los que se ha inducido una sobrexpresión de esta molécula se presentan exacerbaciones de colitis letales (10).
Otra citoquina que tiene relevancia en la patogénesis de EII es la IL-1. Dicha molécula actúa como promotor de la respuesta inmune, el sistema de IL-1 es regulado por el receptor antagonista IL-1Ra así, la actividad biológica de esta citoquina queda determinada por la relación IL-1Ra/IL-1. En pacientes afectados por EII esta relación es inversamente proporcional a la actividad de EII, por lo que la insuficiente producción de IL-1Ra en pacientes con EII podría ser un factor involucrado en la patogénesis de la enfermedad (8,10).
Existen otras citoquinas como la IL-6 , cuyo receptor agonista soluble, tiene concentraciones circulantes elevadas en EII en contraste con pacientes sanos y otras formas de colitis (8,10).
Se menciona en la literatura que tanto en EC como en CU, existen fallas en el sistema regulador de la apoptosis, lo que determina que Linfocitos T autoreactivos de la mucosa intestinal no desarrollen el proceso de apoptosis, generando las condiciones para que estas células produzcan una respuesta inflamatoria deletérea para el huésped (6,10).
Factores Neurohumorales : Existen reportes en la literatura que relacionan la ausencia de estrés psico-social con un aumento del tono parasimpático a nivel intestinal, mediado por acetilcolina, generando una inhibición de la respuesta inflamatoria sistémica al reducir la liberación de TNF-a por parte de los macrófagos. Por el contrario incremento del stress genera estimulación del tono simpático generando en pacientes con CU cambios en la estructura y funcionalidad de la barrera mucosa y el aumento de INF-? (6).
Todas estas alteraciones del sistema inmunitario permiten que la respuesta inmunológica normal en las EII se presente en forma exagerada y desregulada produciendo daño al tejido intestinal y afectando la calidad de vida del paciente.
En resumen:
La epidemiología de las EII ha cambiado en los últimos años, tanto en la población pediátrica como en la población en general. Se percibe un aumento de las tasas de incidencia y prevalencia en zonas geográficas de baja endemia, mientras que en países de alta prevalencia sus valores se han mantenido más bien constantes en el tiempo. Las causas de estas variaciones, se le atribuyen a determinantes ambientales.
El debut de las EII se presenta en general en adultos jóvenes. Sin embargo un porcentaje importante de los casos que va entre un 15 a 25%, debuta durante la edad pediátrica. La edad de presentación promedio, en estudios norteamericanos, se concentra en la adolescencia.
Los factores ambientales juegan un rol en la etiopatogenia de las EII. Existen estudios que relacionan al Tabaco, ACO, AINES, Dieta, Apendicectomia, Stress, Factores Microbiológicos, Factores geográficos y socio-económicos; ya sea en su origen como en el comportamiento clínico de la patología.
La agregación familiar presente en las EII y estudios de concordancia en Gemelos, sugieren la influencia del acervo genético en la patogenia de las EII. A su vez se han relacionado regiones del genoma y a sus proteínas codificadas, como predisponentes a desarrollar una EII como CARD 15/NOD-2 y al gen IBD-5. Así como también, se ha asociado haplotipos con ciertas manifestaciones extraintestinales de este grupo de enfermedades.
Se ha demostrado en tanto en pacientes portadores de EII como en modelos animales, alteraciones del sistema inmunológico. Dichas anormalidades involucran tanto a la inmunidad innata como adaptativa, lo que condiciona una respuesta anómala, ante desafíos inocuos como la microbiota comensal, montando una respuesta inmune sobredimensionada y desregulada, que se traduce en las alteraciones propias de las EII.
Bibliografía
- Figueroa C, Quera R, Valenzuela J, Jensen C. Enfermedades Inflamatorias Intestinales: Experiencia de Dos Centros Chilenos. Revista Médica de Chile 2005, vol. 133: 1295-1304.
- Diefenbach K y Breuer C. Pediatric inflammatory bowel disease. World Journal of Gastroenterology Mayo 2006, vol. 12, n°20: 3204-3212.
- Loftus E. Clinical Epidemiology of Inflammatory Bowel Disease: Incidence, Prevalence, and Environmental Influences. Gastroenterology 2004 vol.126, Nº6: 1504-1517
- Griffiths A. Specificities of inflammatory bowel disease in childhood. Best Practice & Research Clinical Gastroenterology 2004 vol.18, Nº3: 509-523.
- Jae Geun Hyun, Mayer Ll. Mechanisms underlying inflammatory bowel disease. Drug Discovery Today: Disease Mechanisms 2006, vol. 3, No. 4: 457-462.
- Baumgart D, Carding S. Inflammatory bowel disease: cause and immunobiology. The Lancet Mayo 2007 vol.369 Nº 9573: 1627-1640.
- Danese S, Sans M, Fiocchi C. Inflammatory Bowel Disease : The Role of Environmental Factors. Autoimmunity Reviews 2004, vol. 3: 394-400.
- Fiocchi C. Inflammatory Bowel Disease: Etiology and Pathogenesis. Gastroenterology 1998, vol. 115 Nº1:182–205.
- Russell R, Satsangi J. IBD: a family affair. Best Practice & Research Clinical Gastroenterology 2004, vol. 18: 525–539.
- Neuman M. Immune dysfunction in inflammatory bowel disease. Translational Research Abril 2007 Vol.149, Nº4:173-186.
- Ho G, Lees Ch, Satsangi J. Ulcerative Colitis. Medicine Mayo 2007, vol.35, Nº5, Pages 251-296.
- Kim S, Ferry G. Inflammatory Bowel Diseases in Pediatric and Adolescent Patients: Clinical, Therapeutic, and Psychosocial Considerations. Gastroenterology 2004 vol.126, Nº6: 1550-1560
- Suryakanth G, Fiocchi C, Katz J. Inflammatory bowel disease. Best Practice & Research Clinical Gastroenterology 2002 vol.16, Nº1: 77-90.
- Kugathasan S, et al. Epidemiologic and Clinical Characteristics of Children with newly diagnosed inflammatory bowel disease in Wisconsin: A Statewide Population-Based Study. The Journal of Pediatrics, Octubre 2003, vol. 143, Nº4, Pages 525-531.
- Polito J, Childs B, Mellits E, et al. Crohn's disease: Influence of age at diagnosis on site and clinical type of disease. Gastroenterology 1996, vol. 111: 580–586.
- Griffiths A, Harris K, Smith C et al. Prevalence of inflammatory bowel disease in first-degree relatives of children with IBD. Gastroenterology 1997, vol. 112: A2913.
- Peltekova V, et al. Functional variants of OCTN cation transporter genes are associated with Crohn's disease. Nat. Genet. 2004, vol. 36: 471–475.
- Hendrickson et al. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. Enero 2002, vol.15, Nº1:79-94.
- Kanauchi O, Mitsuyama K, Araki Y, Andoh A. Modification of intestinal flora in the treatment of inflammatory bowel disease. Curr Pharm Des 2003, vol. 9: 333–346.
- Hugot JP, Alberti C, Berrebi D, Bingen E, Cezard JP. Crohn's disease: the cold chain hypothesis. Lancet 2003, vol. 362: 2012–2015.
- Agius L. A primary dysregulation in the immunoregulatory role of the intestinal mucosal epithelial cell in inflammatory bowel disease pathogenesis? Biology of inflammatory response as tissue pattern entities in Crohn's versus ulcerative colitis. Journal of Theoretical Biology 2004, vol. 227: 219–228.
|