Introducción
El Neuroblastoma es uno de los tumores sólidos malignos más frecuentes en los niños. Se origina de la cresta neural, durante la embriogénesis, y puede aparecer en cualquiera de los sitios anatómicos a lo largo de la cadena ganglionar simpática desde el cuello a la pelvis, así como en la glándula suprarrenal. Es el tumor más frecuente en el periodo de lactante. El 95% de los casos se diagnostica antes de los 10 años de edad y el 90% ocurre en menores de 5 años.
La incidencia del neuroblastoma varía en las distintas regiones del mundo y también en relación a los grupos etarios. En la casuística del grupo cooperativo chileno Programa Infantil de Drogas Antineoplásicas (PINDA) el neuroblastoma representa el 4,2% de las neoplasias de la infancia. En EEUU la prevalencia estimada es de 1 caso por 8.000 a 10.000 recién nacidos vivos, diagnosticándose 500 casos nuevos cada año.
El origen del Neuroblastoma fue postulado por primera vez en 1964 por Virchow, en Berlín. El nombre de neuroblastoma fue propuesto por James Homer Wright en 1910. En 1916, William Barlett, en Missouri, logró la primera extirpación exitosa en un niño que sobrevivió por 15 años. Cushing y Worbach reportaron en 1927, en una revisión de 10 años, la maduración de neuroblastoma a ganglioneuroblastoma. En 1928, Colmes y Dresser usaron radioterapia como tratamiento de neuroblastoma sin éxito. Los agentes citotóxicos comenzaron a ser usados en 1940 sin resultados hasta 1965 en que James et al. (St. Jude Children’s Hospital) parecen tener pequeños efectos en la sobrevida de estos pacientes con la quimioterapia combinada.
A diferencia de lo sucedido con otros tumores embrionarios de niños, como leucemia, rabdiomiosarcoma, tumor de Wilms, linfoma y hepatoblastoma, en los que gracias al tratamiento combinado de las distintas modalidades terapéuticas del cáncer la sobrevida ha aumentado sostenida y considerablemente, el pronóstico en tumores neuroblásticos sigue siendo incierto. Esta se debe a que este tumor muestra una marcada heterogenicidad biológica que se manifiesta en un comportamiento y evolución muy variable, según un gran grupo de características dependientes de cada paciente en forma individual. El pronóstico es influido por esta variedad de factores en los que debe incluirse la edad. (1-4) En el 95% de los casos de neuroblastoma se presenta en menores de 10 años de edad. El 50% se diagnostica antes de los 2 años y más de 2/3 durante los primeros 5 años.
Se ha observado que existen 2 tipos de neuroblastoma con pronósticos muy distintos, los cuales no se diferencian histológica ni bioquímicamente, pero si desde el punto de vista genético-molecular, pudiéndose determinar factores de riesgo desfavorables: Nmyc amplificado, deleción de 1p, DNA euploide, falta de expresión de CD44 y N-ras. Estos factores de riesgo rara vez se presentan en menores de 1 año, pero sí están presentes en 1/3 de los neuroblastomas etapa 3 y 4 de niños mayores de 1 año.
Actualmente el Nmyc es considerado el mejor parámetro genético-molecular para definir grupos de riesgo. Por algunos años la edad del paciente y la etapa de la enfermedad en el momento del diagnóstico fueron las 2 variables más determinantes en el pronóstico del neuroblastoma. Evans encontró que los lactantes menores de 1 año de vida con estadios I, II y IV-s tenían significativamente mejor pronóstico. Los pacientes mayores de 1 año y aquellos con estadios más avanzados, o sea, III y IV eran de peor pronóstico. El peor pronóstico de sobrevida se observa en pacientes mayores de 1 año, en estadio IV y metástasis óseas corticales. El sitio de tumor primario también fue considerado predictivo de sobrevida, según algunos investigadores. (5) Pacientes con tumores en cuello, pelvis y mediastino tenían mejor pronóstico comparados con aquellos que presentaban tumores en el retroperitoneo (paraespinal o suprarrenal).
Objetivo
El objetivo de este trabajo fue determinar si existe relación entre la forma inicial de presentación clínica de aquellos pacientes en etapa IV con compromiso óseo y las posibilidades de tratamiento quirúrgico primario. Además, determinar la relación entre la localización del tumor primario y el pronóstico.
Material y métodos
Se realizó una revisión retrospectiva de 22 fichas de pacientes tratados por neuroblastoma en el Hospital Roberto del Río entre 1994–2004.
Los datos se analizaron estadísticamente mediante pruebas Fisher- Irwing. Se consideraron para el estudio los siguientes datos: frecuencia por sexo y edad, presentación inicial, localización, diagnóstico inicial, tiempo de evolución, etapa al diagnóstico, histopatología y el tratamiento inicial aplicado.
Resultados
Se encontró que el 50% (11) de los casos correspondían a niños de 1 a 5 años, 27% (6) a menores de 1 año y 22% (5) a niños entre 5 a 7 años. Se presentó en igual número de mujeres que de hombres.
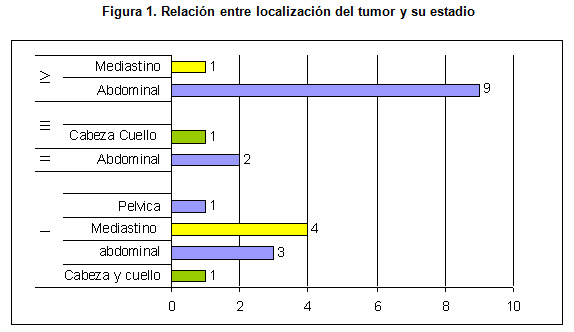
La forma de presentación clínica más frecuente fue la de aquellos tumores abdominales y pélvicos, produciéndose con mayor frecuencia diarrea. También se puede presentar con dolor, masa o distensión abdominal. La segunda forma de presentación más frecuente fue la de neuroblastoma en mediastino, pudiendo manifestarse como un síndrome bronquial obstructivo más comúnmente, asimismo como disnea o masa mediastínica. (Figura 1.)
Los diagnósticos diferenciales más frecuentemente planteados, en un principio, frente a un neuroblastoma fueron: tumores de otro origen (36% de los casos); enfermedad osteoarticular (18% de los casos); menos comúnmente maltrato infantil y ganglioneuroma. El diagnóstico inicial fue el correcto en el 27% de los casos.
La ubicación más habitual de los tumores en esta revisión fue la abdominal (63%), la sigue en frecuencia los tumores en mediastino (23%), y luego los tumores pélvicos (5%); en el 9% de los casos el tumor primario tuvo otras localizaciones.
En relación al tiempo de evolución previo al diagnóstico, 59% (13) de los pacientes tenía menos de 1 mes de clínica, 32% (7) de los casos tenían entre 2 a 7 meses de evolución y sólo el 9% (2) tenía más de 1 año.
La mayoría de los pacientes, al momento del diagnóstico, se encontraban en etapa I o en etapa IV (45% de los casos para cada etapa).
De los casos que se diagnosticaron en la etapa I, la mayoría de los neuroblastomas se hallaban ubicados en mediastino. De los casos que se diagnosticaron que se encontraban en etapa IV, la mayoría tenían localización abdominal. (Figura 1)
Con respecto a la clasificación histopatológica, 11 de los tumores se tipificaron como neuroblastoma; 10 como ganglioneuroblastoma y sólo 1 caso de ganglioneuroma.
Para la confirmación diagnóstica y etapificación de la enfermedad se practicaron exámenes imagenológicos (tomografía axial computarizada, ecotomografía y cintigrafía) y marcadores genético-moleculares (SPOT, VAM, MIBG y Nmyc).
Como tratamiento inicial a la mayoría de los casos, 36% (8), se le efectuó una biopsia del tumor; con similar frecuencia se realizó resección total del tumor como primer tratamiento (32%); en 4 casos se decidió comenzar tratamiento con quimioterapia y a 3 casos se le practicó resección parcial del tumor en primera instancia.
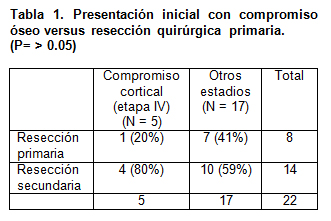
No existe una relación estadísticamente significativa (p=>0.05) entre aquellos casos que tenían neuroblastoma en etapa IV con metástasis óseas corticales, y efectuar la resección primaria del tumor. (Tabla 1)
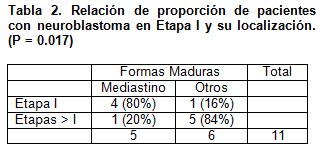
Se demostró una relación estadísticamente significativa (p=<0.05) entre las formas de presentación maduras del neuroblastoma y su localización. Es decir, de los tumores ubicados en el mediastino, todos se informaron como formas maduras.
Además se encontró que existe una relación entre pacientes con neuroblastoma en estadío I y localización mediastinal, lo que no ocurrió en tumores con otras ubicaciones. (p=<0.05) (Tabla 2)
Conclusiones
- El compromiso óseo cortical implica estadio IV de la enfermedad. Sin embargo no podemos deducir que existe una relación entre estadio IV con compromiso óseo e imposibilidad de tratamiento quirúrgico primario (p >0.05), si comparamos con otras localizaciones.
- Las formas maduras son más frecuentes en la localización mediastínica (p < 0.05).
- La frecuencia de pacientes con formas maduras de neuroblastoma en etapa I es mayor en localización mediastínica que en otras. (p = 0.017)
- Existiría una relación entre localización mediastínica y un mejor pronóstico de la enfermedad.
- Consideramos que en este estudio obtuvimos resultados válidos importantes, pero sabemos que el número de casos analizados era escaso y que tal vez con un número mayor podrían demostrarse otras relaciones interesantes, incluyendo la primera hipótesis de este trabajo.
Discusión
Aunque la información acumulada es considerable alrededor del comportamiento del neuroblastoma, el pronóstico sigue siendo incierto (6). Este tumor maligno, tan común en los niños, sigue siendo un enigma debido a la gran variabilidad de su conducta. Información obtenida de seguimientos en fetos y de tamizaje en lactantes demuestran claramente que el neuroblastoma puede regresar espontáneamente.
Los estudios histopatológicos han clarificado que algunos neuroblastomas son tumores más inmaduros e indiferenciados que otros, y que los primeros muestran una evolución progresiva y fatal.
Actualmente el pronóstico es mejor en los lactantes menores de 1 año de edad, en pacientes con tumor localizado que pueden ser resecados en su totalidad (estadios I y II), en pacientes con histología Shimada favorable y factores biológicos de riesgo, y en pacientes en etapa IV-s. Casos de riesgo intermedio incluyen: estadio III (INSS), sin factores de riesgo biológicos y estadio IV en pacientes menores de 1 año sin factores biológicos de riesgo. (7-10)
El área en que continúan los problemas y requieren especial atención es aquella relacionada con los niños mayores de 1 año, pacientes con estadios avanzados de la enfermedad (III y IV) y pacientes con variables biológicas con pronóstico adverso. También deben mejorarse la prevención y tratamiento de las complicaciones por terapia agresiva multimodal (daño por irradiación corporal y total, inmunosupresión, escoliosis, efectos en desarrollo y crecimiento, maduración sexual y segundo cáncer). (11, 12) El reconocimiento de características biológicas y genéticas pueden categorizar al tumor en grupo de riesgos (alto, intermedio y bajo) que determinen los futuros protocolos de tratamiento. (13) Es necesaria una mejor comprensión de los factores que influencian la regresión y diferenciación tumoral (eventos genéticos, embriológicos y teratogénicos) y la relación huésped-tumor. En niños con factores biológicos de alto riesgo es necesario desarrollar anticuerpos monoclonales contra el tumor y nuevas técnicas de limpieza de células tumorales de médula ósea y nuevas y más efectivas drogas quimioterápicas (con o sin irradiación corporal total). Están siendo evaluados con la esperanza de mejorar el pronóstico del neuroblastoma avanzado; el transplante de médula ósea con mejores alternativas de rescate hematológico, el uso de factores de crecimiento y de moduladores tumorales (ácido cis-retinoico) que promueven la regresión y diferenciación. (14, 15)
Referencias
- Look A. et al. Clinical Relevante of tumor cell ploidy and N-myc gen amplification in chilhood neuroblastoma: a pediatric oncology group study. J. Clin. Oncol. 1991; 9: 581.
- Altman A., Baehner R. Favorable prognosis for survival in children with coincident opsomyoclonus and neuroblastoma. Cancer. 1976; 37: 846.
- Caron H. et al. Allelic loss of chromosome Ip as predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 1996; 334: 225.
- Coldman A. et al. Neuroblastoma influence of age at diagnoses, stage, site and sex on prognosis. Cancer. 1980; 46: 1896.
- Filler R. et al. Favorable outlook for children with mediastinal neuroblastoma. J. Pediatr. Surg. 1972; 7: 136.
- Rostion C.G. Cirugía pediátrica. Santiago- Chile, Mediterráneo, 2000.
- Grosfeld J. et al. Neuroblastom in the first year of life: Clinical and biologic factors influencing outcome. Semin. Pediatr. Surg 1993; 2: 37.
- Grosfeld J. et al. Metastasic neuroblastoma: factors influencing survival. J. Pediatr. Surg. 1978; 13: 59.
- Haase G. et al. Aggressive surgery combined with intensive chemotherapy imrpoves survival in poor risk neuroblastoma. J. Pediatr. Surg. 1991; 26: 1119.
- Jennings R. et al. Fetal neuroblastoma: prenatal diagnoses and natural history. J. Pediatr. Surg. 1993; 28: 1168.
- LaQuaglia M. et al. Stage IV neuroblastoma: diagnosis at more than one year, gross total resection and clinical outcome. J. Pediatr. Surg. 1994; 29: 1163.
- Ninane J. et al. Stage II neuroblastoma adverse prognostic significance of lymph node involvement. Arch. Dis. Child. 1982; 57: 438.
- Shimada H. et al. Histopathologic prognostic factors in neuroblastomas: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastoma. J. Natl. Cancer Inst 73: 405, 1984.
- Shorter N. et al. The role of surgery in the management of stage IV neuroblastoma: a single institution study. Med. Peditr. Oncol. 1995; 24: 287.
- Meitar D. et al. Tumor angiogenesis correlated with metastasic disease, N-myc amplification and poor outcome in human neuroblastoma. J Clin. Oncol 1996; 14: 405.