Introducción
La mayoría de los niños con circunferencia craneana (CC) superior al promedio normal para su edad y sexo no tienen patología grave, sin embargo el médico debe considerar siempre la posibilidad de un cuadro evolutivo dentro de los cuales el más frecuente e importante a descartar es la hidrocefalia. El enfrentamiento inicial va a depender de la edad del niño, del perfil temporal del crecimiento de la cabeza y de la presencia de síntomas o signos indicativos de patología neurológica. Un análisis cuidadoso de los datos obtenidos a través de la anamnesis y del examen físico/neurológico, y una adecuada valoración del desarrollo psicomotor permitirán definir las probables causas de la macrocefalia y la necesidad de exámenes complementarios, evitando realizar procedimientos innecesarios.
La medición y registro del perímetro del cráneo en cada control del niño, especialmente en los primeros 2 años de vida es crucial para evaluar adecuadamente variaciones en el tamaño craneal y para la detección precoz de hidrocefalia.
El tamaño del cráneo está determinado por el encéfalo, líquido céfalo-raquídeo (LCR), sangre y el grosor de los huesos (1). Su incremento refleja el crecimiento del encéfalo, de ahí la importancia de su medición, registro y valoración con las tablas de CC en cada control pediátrico. Los recién nacidos de término (RNT) presentan un perímetro craneano que oscila entre 35 y 36 cm con un crecimiento aproximado de 12 cm durante el primer año de vida (1) siendo la CC de las niñas levemente menor que la de los varones. El incremento de la CC en el primer trimestre es más acelerado, en promedio de 2 cm/mes, disminuye durante el segundo trimestre a 1 cm/mes y en el tercer y cuarto trimestre a 0,5 cm/mes (1). En los recién nacidos de pretérmino (RNPT), la CC debe ser corregida hasta los 18 meses de acuerdo a edad gestacional, recomendándose el uso de tablas adaptadas como la de Fenton (1). En el recién nacido algunas condiciones pueden alterar la medición de la CC, como la bolsa serosanguínea, cefalohematoma (1), hemorragia subgaleal, cabalgamiento de los huesos parietales o fracturas craneales.
Medición de CC
La medición de la CC con técnica correcta se realiza con la cabeza en el plano de Frankfort (línea imaginaria que pasa por el borde inferior de la órbita y el punto más alto del conducto auditivo externo que deben estar paralelos al suelo), rodeando la cabeza con una cinta métrica por encima del arco supraciliar y por sobre la protuberancia occipital externa. Se deben realizar dos o tres mediciones para reducir el error y registrar el perímetro máximo (1,6). La Academia Americana de Pediatría recomienda que la medición de la CC debe realizarse en cada control de salud del niño, al menos hasta los 2 años, sin embargo, debemos considerar que el crecimiento craneano continúa hasta los 5 años, momento en el que prácticamente se estaciona (2). La confección de una curva de crecimiento permitirá la detección de alteraciones, ya sea una medición aislada fuera de la curva normal o un cambio en el carril de crecimiento respecto de mediciones previas, las que deben ser detectadas tempranamente.
Las alteraciones en el crecimiento del cráneo pueden corresponder a una variante normal, pero implican un desafío diagnóstico debido a las múltiples causas, congénitas (genéticas o embriopáticas) o adquiridas que deben descartarse.
Definición de macrocefalia
La macrocefalia se define como una mediciónde la CCmayor de dos desviaciones estándar (DS) por sobre la media de una determinada edad, sexo y tiempo de gestación (1, 2, 7). Su prevalencia es de 5% en la población general (8).
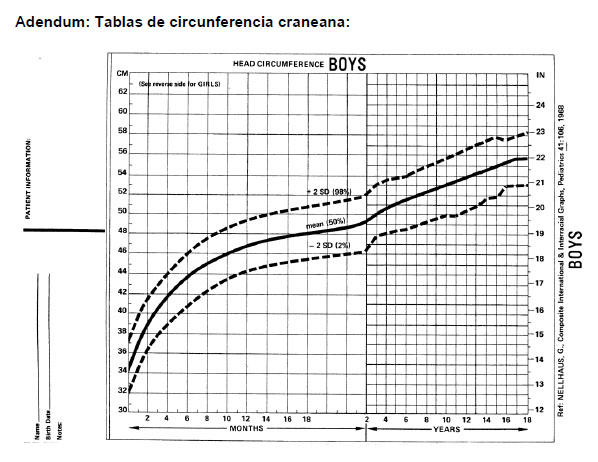
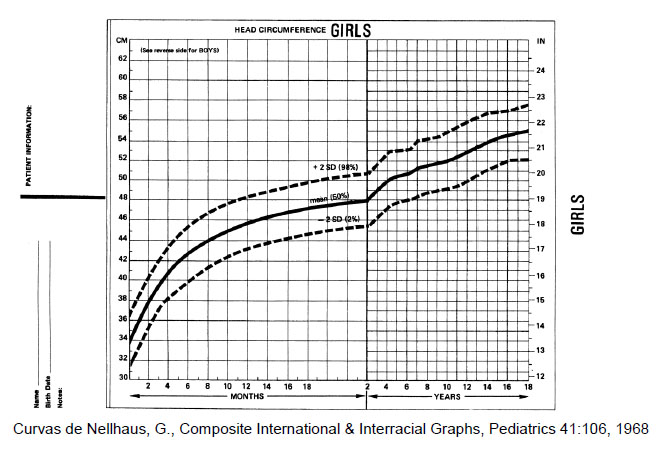
Existen diversas curvas de crecimiento craneano. Entre las más usadas están las curvas de Nellhaus (9) (Adendum), del Centro Nacional de Salud y Estadísticas (NCHS de su sigla en inglés), del Centro para el Control y Prevención de Enfermedades (EEUU) y las de la Organización Mundial de la Salud, las que difieren entre si sobre todo en los percentiles superiores, por lo que se ha planteado realizar más investigaciones clínicas para obtener mejores curvas (10). En la Atención Primaria de Salud (APS) de nuestro país, se utiliza la curva NCHS (11).
El diagnóstico de macrocefalia es relevante dado que puede ser la primera manifestación de trastornos neurológicos potencialmente graves, como una hidrocefalia. En centros de derivación de niños con alteraciones del desarrollo se encontró que el 1,4% presentaba macrocefalia, siendo la hidrocefalia la causa de ésta en un tercio de los casos (12). En niños con trastornos del desarrollo, la asociación con macrocefalia constituye un factor de riesgo de crisis febriles y epilepsia (12).
Clasificación y etiología
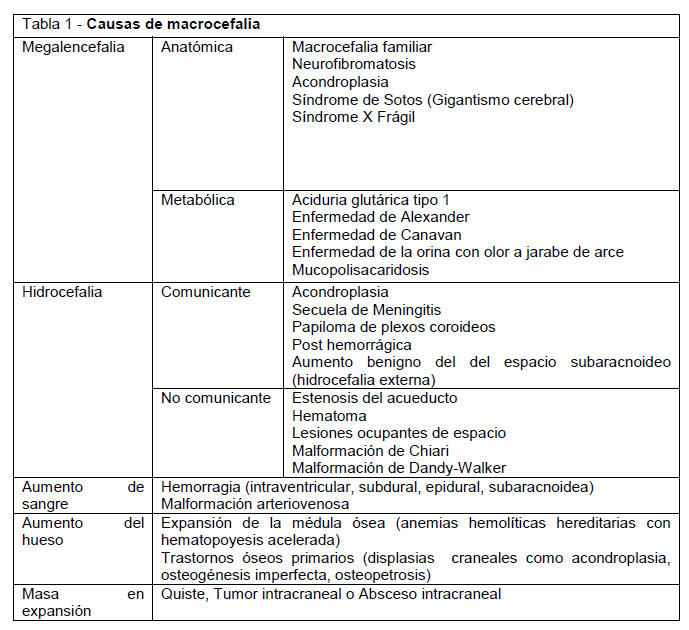
Existen múltiples clasificaciones de macrocefalia: de acuerdo a su etiología (congénita y adquirida) (7), a su evolución (macrocefalia progresiva o no progresiva) (13), características clínicas (hipertensiva o normotensiva) o según el espacio que se encuentra aumentado (LCR o encéfalo). Se denomina megalencefalia al aumento del tamaño del encéfalo e hidrocefalia al aumento del LCR intracraneal, para diferenciar del engrosamiento de los huesos del cráneo y hemorragias en los espacios subdural o epidural (1,3,13). La megalencefalia puede tener causas anatómicas o metabólicas y la hidrocefalia se clasifica en comunicante y no comunicante, según si se mantiene o no comunicación entre ventrículos y espacio subaracnoideo respectivamente. (Tabla 1).
Megalencefalia
Corresponde a un aumento del volumen del parénquima cerebral sin signos de hipertensión intracraneal, aumento del LCR o lesiones ocupantes de espacio (13).
En modelos animales se han demostrado el rol que juega el factor de crecimiento IGF-1 en el aumento del número de neuronas (14) y el rol de las proteínas CD81 en el aumento del número de astrocitos (15). Otros modelos han encontrado mutaciones que determinan crecimiento del tamaño del encéfalo en genes proapoptóticos como el del receptor EphA7 (16).
Megalencefalia genética (anatómica)
La megalencefalia anatómica se refiere al aumento del tamaño del cerebro a expensas del aumento del tamaño o número de las células, sin una enfermedad metabólica de base (1). La aproximación inicial se dirige a pesquisa o descarte de
anormalidades del examen neurológico y desarrollo, que permite definir 2 grandes grupos (niños sanos y niños con alteraciones):
1 - Megalencefalia familiar benigna
La causa más frecuente de macrocefalia, en diversas series publicadas, es la megalencefalia familiar benigna, la cual se diagnostica en niños sin déficit neurológico y con familiares con macrocefalia (13).
La CC se encuentra de 2 a 4 cm por sobre las 2DS, con tamaño corporal y desarrollo psicomotor normal, sin alteraciones neurológicas y sin características que sugieran un síndrome específico. Se presenta en aproximadamente el 2% de la población. La CC puede ser normal al nacimiento y luego acelera su crecimiento. Característicamente alguno de los padres presenta un tamaño aumentado de la cabeza. Se ha propuesto herencia autosómica dominante, pero es posible que sea multifactorial (1,13). La Tomografía computada (TC) es normal.
2 - Megalencefalia asociada a anormalidad neurológica
Si la megalencefalia se asocia a alteración neurológica, debe investigarse si ésta es estacionaria o progresiva. Los síntomas neurológicos son variables e incluyen déficit cognitivo y/o de atención, retraso del lenguaje, trastornos de aprendizaje, déficit motor, crisis epilépticas y ocasionalmente dismorfias menores como sindactilia. Es importante diagnosticar los síndromes neurocutáneos, especialmente la neurofibromatosis, para una oportuna pesquisa de complicaciones. La TC y la Resonancia Magnética (RM) pueden presentar alteraciones como leve dilatación ventricular y agenesia del cuerpo calloso (1,13).
3 - Megalencefalia con alteraciones genéticas
La megalencefalia puede estar presente en algunos trastornos genéticos, como en el síndrome de X frágil, causa más frecuente de discapacidad intelectual hereditaria con una prevalencia de 1 en 1200 varones y 1 en 2500 mujeres. En la resonancia magnética de cerebro se aprecia una disminución de la relación sustancia blanca-gris, núcleo caudado amplio y disminución del giro temporal superior y del vermis cerebeloso, con aumento relativo del IV ventrículo (17).
4 - Megalencefalia en síndromes genéticos no cromosómicos
Síndrome de Sotos. Denominado también “Gigantismo Cerebral” tiene incidencia de 1 en 10.000 a 1 en 30.000 nacimientos (18). Los niños afectados presentan una CC normal al nacer, que aumenta aceleradamente hasta los 3 años, estabilizando posteriormente su tamaño. Tienen dismorfias típicas como cara alargada, frente y mandíbula prominente, escaso pelo fronto-parietal, fisuras palpebrales oblicuas hacia caudal, hipertelorismo aparente, erupción prematura de dientes, manos y pies grandes, cardiopatías congénitas, cataratas y glaucoma, y presentan mayor riesgo de neoplasias (tumor de Wilms, carcinoma hepatocelular, neuroblastoma, etc.). Pueden presentar además hipotonía, retraso en el lenguaje y déficit cognitivo variable. La TC puede evidenciar ventrículomegalia, anomalías del cuerpo calloso y prominencia del cuernos occipitales (1,17).
Síndrome de Weaver: fenotípicamente se asemeja al síndrome de Sotos, pero el patrón de crecimiento es diferente, presentando macrocefalia en la adultez. Las dismorfias incluyen pulgares anchos, pulpejos prominentes, pliegues plantares, clinodactilia, extensión limitada del codo y rodilla, alteraciones de vértebras cervicales, llanto ronco, mayor riesgo de neoplasias y retraso motor con hipertonía. Habitualmente no presentan déficit cognitivo. Las imágenes pueden mostrar quistes del septo pelúcido, paquigiria e hipervascularización (17).
Otros síndromes genéticos de más reciente descripción asociados a mutaciones de un gen supresor de tumores involucrado en un número creciente de cánceres, el gen PTEN (del inglés, PHOSPHATASE AND TENSIN HOMOLOG, gen homólogo de la fosfatasa y angiotensina), localizado en el locus 10q23.31, que se ha descrito como causante de varios síndromes genéticos cuya manifestación central es la macrocefalia asociado en la mayoría de los casos a sobrecrecimiento pondoestatural y neoplasias. Mutaciones en este gen se han demostrado en casos de síndromes
conocidos: Síndrome de Bannayan-Riley-Ruvalcaba, Síndrome de Lhermitte-Duclos, Síndrome de Macrocefalia/autismo, Síndrome de hamartoma/tumores asociado a PTEN, Síndrome de Cowden y Síndrome de VATER con macrocefalia y ventriculomegalia. La importancia de reconocer estos cuadros radica en que requiere seguimiento estricto a la largo de la vida realizando estudios para pesquisa precoz de neoplasias (19).
Acondroplasia: presenta transmisión autosómica dominante con 80% de mutaciones nuevas. Se caracteriza por talla baja (enanismo), acortamiento de extremidades, frente prominente, hipoplasia media de la cara y limitación en la extensión de codos. Además presentan estenosis del canal raquídeo a nivel dorsal y lumbar (20). La TC muestra una fosa posterior pequeña y aumento de los senos esfenoidales (1).
5 - Megalencefalia en síndromes neurocutáneos
Los síndromes neurocutáneos son trastornos que afectan múltiples sistemas, incluyendo sistema nervioso y piel, por su origen embriológico común. Las manifestaciones cutáneas pueden estar presentes desde el nacimiento o aparecer durante el primer año. En este grupo encontramos Neurofibromatosis tipo 1 (NF1), un trastorno multisistémico con prevalencia de 1 en 3500.Sus características incluyen manchas café con leche de la piel, efélides axilares o inguinales, neurofibromas cutáneos, glioma del nervio óptico, hamartomas del iris (nódulos de Lisch), lesiones óseas y el antecedente de un familiar afectado en primer grado (21).
Megalencefalia Metabólica
La megalencefalia puede ocurrir en enfermedades de depósito o en trastornos del metabolismo intermediario, debido a depósito de metabolitos anómalos o edema cerebral. En general, estos cuadros se presentan con regresión del desarrollo psicomotriz y con crisis epilépticas (1). Las causas son múltiples (Tabla 2), y deben ser sospechadas en pacientes con macrocefalia y deterioro clínico (regresión del desarrollo psicomotor), así como en macrocefalias progresivas.
Hidrocefalia
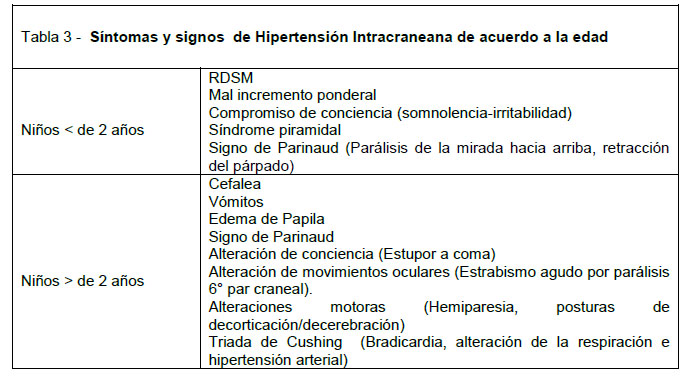
La hidrocefalia se define como un aumento del LCR intracraneal, observándose en las imágenes aumento del tamaño ventricular y/o del espacio subaracnoideo. Se clasifica en hidrocefalia comunicante y no comunicante dependiendo de la permeabilidad entre el sistema ventricular y el espacio subaracnoideo. La hidrocefalia es la principal causa de macrocefalia al nacer, por lo que su detección y tratamiento precoz es crucial. En un niño con hidrocefalia lo prioritario es la pesquisa de hipertensión intracraneana, que constituye una urgencia neuroquirúrgica. La hipertensión intracraneana presenta distinta sintomatología dependiente del cierre o no de las suturas (Tabla 3). El tratamiento de la hidrocefalia implica generalmente la instalación de una derivativa o válvula que permita evacuar el LCR desde el sistema ventricular a peritoneo o atrio, lo que resuelve esta emergencia; sin embargo debe tenerse en cuenta que todo paciente que porta una derivativa puede presentar a lo largo de su vida nuevos episodios de hipertensión intracraneana producidos por disfunción valvular. La disfunción de una derivativa debe sospecharse en todo paciente con antecedentes de hidrocefalia derivada que presenta cefalea aguda, hiperaguda o progresiva.
Niños macrocefálicos debido a hidrocefalia, tienen mayor frecuencia de cesáreas y de eventos adversos perinatales como hemorragia intracraneal y convulsiones neonatales (12).
Hidrocefalia comunicante
Se produce por aumento en la producción o disminución en la reabsorción del LCR (1,13).
1 -Aumento de la producción
Es poco frecuente puesto que la reabsorción del LCR supera por mucho a la producción de éste. Un ejemplo clásico que cursa con elevada producción de LCR es el papiloma del plexo coroideo, sin embargo, en algunos casos puede producirse obstrucción del agujero de Monro, lo que la transforma en una hidrocefalia no comunicante (1,13).
2 - Disminución de la absorción
Se produce habitualmente por disfunción de la reabsorción del LCR a nivel de las vellosidades aracnoidales debido a múltiples causas: infecciones, hemorragia,
infiltración tumoral de meninges y espacio subaracnoideo en leucemias, linfomas y metástasis, post meningitis o post hemorragia relacionada a bloqueo del espacio subaracnoideo por residuos proteicos, o post trombosis venosas en que se altera la absorción de LCR por obstrucción de los senos venosos de la duramadre (1,13).
3 - Malformativas
La hidranencefalia es una condición congénita en la que los hemisferios cerebrales son sustituidos por quistes de paredes finas llenos de LCR, considerándose una forma extrema de porencefalia. La causa de la hidranencefalia no es bien conocida, se ha relacionado a una anomalía del desarrollo del cerebro o más frecuentemente a una destrucción del tejido cerebral por injuria isquémica de territorio carotideo en la etapa intrauterina, con atresia del acueducto de Silvio que genera hipertensión intracraneana. La hidrocefalia obstructiva progresiva, si no es tratada oportunamente, puede causar un cuadro muy similar a hidranencefalia. El diagnóstico de la hidranencefalia se realiza con RM que muestra ausencia de hemisferios cerebrales (22).
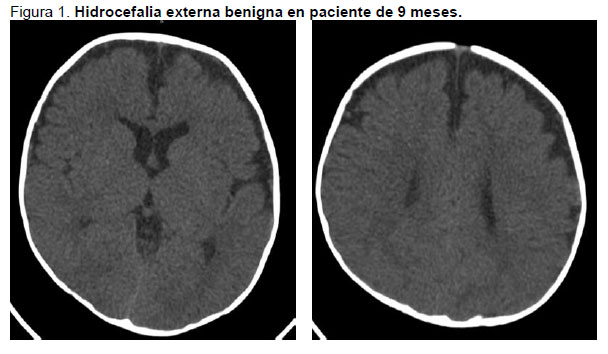
4 - Hidrocefalia externa benigna
La hidrocefalia externa benigna, llamada también espacio subaracnoideo complaciente, aumento benigno del tamaño del espacio subaracnoideo o colección peri cerebral benigna, corresponde al aumento del LCR en el espacio subaracnoideo con ventrículos normales o levemente aumentados de tamaño. (Figura 1). Es una de las causas más frecuentes de macrocefalia en el niño junto con la megalencefalia familiar, solo distinguibles mediante neuroimágenes.
Es un cuadro de etiología y fisiopatología controvertida, que afecta a lactantes y ocasionalmente a recién nacidos, cursando con incremento rápido de la CC. En las neuroimágenes se observa un espacio subaracnoideo amplio especialmente a nivel de lóbulos frontales, y leve aumento del tamaño ventricular que habitualmente se resuelve espontáneamente hacia los 2 años de edad (23, 24). (Figura 1) El examen neurológico es habitualmente normal, aunque puede cursar con adquisición más lenta de hitos del DSM o retraso del DSM “transitorio”. En centros de neurología pediátricos se ha comunicado un 0,6% de hidrocefalia externa benigna como hallazgo en neuroimágenes, siendo 2/3 varones. El 40% de estos niños tienen al menos un familiar con macrocefalia, por lo que se ha planteado un modelo de herencia multifactorial. Una teoría sugiere que las vellosidades aracnoidales son incapaces de absorber el LCR producido debido a inmadurez. El tratamiento quirúrgico propuesto en algunos casos con buen resultado, no está exento de riesgos. (25, 26).
Hidrocefalia no comunicante
Es la causa más frecuente de hidrocefalia neonatal, en la cual existe dilatación de los ventrículos por obstrucción de la salida del LCR desde el sistema ventricular hacia el espacio subaracnoideo (1,13).
Entre las causas se pueden encontrar:
1 - Estenosis del acueducto de Silvio: El diámetro del acueducto de Silvio al nacimiento es de 0,5 mm en promedio, por lo que su compresión por tumores o hemorragias puede obstruirlo fácilmente. Aproximadamente 5% de los casos de hidrocefalias congénitas de curso severo corresponden a atresia o estenosis congénita del acueducto de Silvio, frecuentemente de causa genética por mutaciones en el gen de la molécula de adhesión neural L1CAM ubicado en locus Xq28, que es esencial en el desarrollo neurológico (27). La CC puede alcanzar hasta 50 cm al nacimiento, con grave desproporción cefalopélvica. El diagnóstico debe efectuarse en período prenatal por ecografía. El 50% de estos pacientes presenta el pulgar aducido e hipoplásico lo que orienta al diagnóstico, y un porcentaje menor agenesia del cuerpo calloso, paquigiria, polimicrogiria o ausencia de parte del haz piramidal (28).
2 - Anomalía de Chiari: Se caracteriza por descenso del cerebelo y tronco encefálico a través del agujero magno. Puede asociarse a anomalías del tronco encefálico o a distintos niveles del neuroeje.
3 - Síndrome de Dandy-Walker: Corresponde a una obstrucción de los agujeros de Luschka y Magendie, asociada a agenesia de vermis cerebeloso posterior, megacisterna magna y dilatación quística
del IV ventrículo. La hidrocefalia es tardía. Este cuadro puede ser diagnosticado prenatalmente con ecografía (29).
4 - Síndrome de Klippel-Feil: Existe fusión anormal de vértebras cervicales con alteración de la porción inferior del tronco y obstrucción a la salida del LCR a nivel del IV ventrículo. Se manifiesta por implantación baja del pelo, dificultad en movimientos cervicales, escoliosis y movimientos en espejo de las manos. Su diagnóstico se establece mediante radiografías, TC y RM cervical, que muestran esta anormalidad (30), en ocasiones asociada a malformación de Chiari.
Lesiones con efecto masa
Las lesiones con efecto de masa incluyen tumores (astrocitomas, meduloblastomas o tumores de plexo coroideo), hemorragias, hematomas, abscesos o infecciones granulomatosas. En general producen hidrocefalia subaguda y el diagnostico se establece mediante TC o RM de cerebro.
Engrosamiento óseo
Característicamente la CC es normal al nacimiento. Entre las causas a considerar se encuentran: anemia crónica, displasias óseas, osteogénesis imperfecta, raquitismo, síndrome de Silver-Russell, hiperfosfatemia. (1,13)
Evaluación del niño con macrocefalia
La evaluación se inicia con una detallada anamnesis en la que debe incluirse registro de mediciones en la curva de crecimiento de la CC, desarrollo psicomotor, búsqueda dirigida de síntomas de hipertensión intracraneana (Tabla 3) y crisis epilépticas. En la anamnesis remota es importante investigar antecedentes prenatales como uso de drogas, exposición a radiaciones o a tóxicos, posibles infecciones congénitas y resultados de ecografías obstétricas. El estudio imagenológico prenatal puede demostrar la macrocefalia y eventualmente su causa (Síndrome de Dandy-Walker, quistes del plexo coroideo o malformación de la vena de Galeno), pero si la ecografía solo demuestra CC > 2 DS para la edad gestacional, sin otras anormalidades asociadas, no se considera necesariamente factor de riesgo de anormalidad neurológica a largo plazo (29). Es importante buscar antecedentes de parto traumático, cesárea o sufrimiento fetal agudo. También se debe explorar antecedentes familiares de enfermedades neurológicas o macrocefalia.
Examen Físico
Debe realizarse antropometría completa con medición y cálculo de percentiles de peso, talla y especialmente CC en los primeros 2 años de vida para evaluar adecuadamente variaciones de la curva de crecimiento y para la detección precoz de hidrocefalia (31). Evaluar la forma del cráneo y la tensión de las fontanelas. La tensión del bregma debe evaluarse con el niño sentado y tranquilo (sin llanto), siendo normal encontrar una fontanela a nivel o ligeramente deprimida que fluctúa con las fases de la respiración. En el examen segmentario es esencial buscar dismorfias, malformaciones esqueléticas, estigmas cutáneos y visceromegalias.
El examen neurológico debe ser completo incluyendo fondo de ojo y anormalidades que indican hipertensión intracraneana en evolución (Tabla 3) (13).
Exámenes Complementarios: Neuroimágen
En general, se plantea estudio con neuroimágenes en los lactantes con macrocefalia, especialmente en aquellas progresivas, asociadas a retraso del desarrollo o a cualquier anormalidad en el examen físico general o neurológico. La elección de TC o RM debe ser hecha en base al análisis de los elementos anamnésticos y del examen junto a la sospecha de etiologías más probables o aquellas que implican riesgo vital y/o ameritan una conducta terapéutica más urgente.
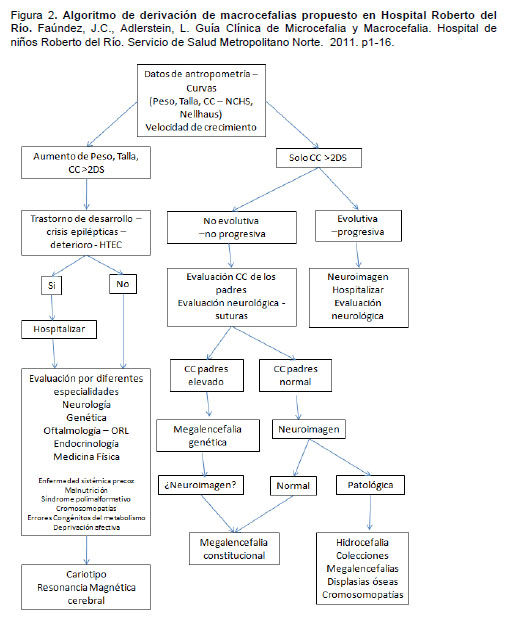
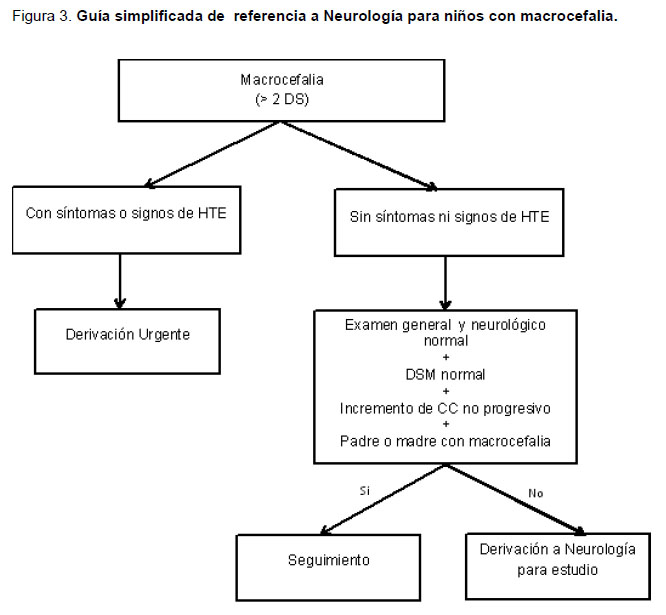
Dos propuestas para referencia de niños con macrocefalia desde atención primaria a especialista se muestran en las figuras 2 y 3, el primero propuesto como guía clínica en el Hospital Roberto del Río (Figura 2 y 3).
Finalmente, si bien en la mayoría de los casos de macrocefalia no se encuentra una causa grave, es fundamental analizar
cuidadosamente la información obtenida en la anamnesis y el examen y junto a una rigurosa evaluación del desarrollo psicomotor a fin de descartar en cuadros que requieran un tratamiento precoz.
Tablas y figuras

Referencias
- Fenichel G. Trastornos en la forma y volumen del cráneo. En: Fenichel, G. Neurología pediátrica clínica. Barcelona-España. Elsevier. 2010. 6° edición, Cap. 18:369-386.
- Fernández E. El examen neurológico. En: Fejerman N., Fernández, E. Neurología Pediátrica. Buenos Aires-Argentina. Editorial Médica Panamericana. 2007. 3° ed., Cap. 2:6-24.
- Schlager G. Alteraciones del tamaño y de la configuración craneana en el lactante. Rev Chil Pediatr.1990; 61:161-165.
- Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr. 2013;14:59.
- Rios A. Microcephaly. Pediatr Rev 1996; 17:386.
- Weiner JS, Lourie JA. Human Biology: A Guide to Field Methods. 1BP No. 9, Blackwell, London. 1969.
- Williams CA, Dagli A, Battaglia A. Research review genetic disorders associated with macrocephaly. American Journal of Medical Genetics Part A. 2008; 146:2023–2037.
- Hamill PV, Drizd TA, Johnson CL, Reed RB, Roche AF, Moore WM. Physical growth: National Center for Health Statistics percentiles. Am J Clin Nutr. 1979; 32:607–629.
- Nellhaus G. Head circumference from birth to eighteen years: practical composite international and interracial graphs. Pediatrics. 1968; 41:106-114.
- Daymont C, Hwang W, Feudtner C, Rubin D. Head-Circumference distribution in a large primary care network differs from CDC and WHO curves .Pediatrics. 2010; 4(126):836-842.
- Hamill PV, Drizd TA, Johnson CL, Reed RB, Roche AF. NCHS growth curves for children birth-18 years. United States. Vital Health Stat 11.1977; 165:i–iv,1–74.
- Nevo Y, Kramer U, Shinnar S, Leitner Y, Fattal-Valevski A, Villa Y, et al. Macrocephaly in Children With Developmental Disabilities. Pediatric Neurology. 2002; 5(27):363-368.
- García J, Romero F. Alteraciones del perímetro craneal: microcefalia y macrocefalia. Pediatr Integral 2003; 8:587-600.
- D’Ercole AJ, Ye P, O’Kusky JR. Mutant mouse models of insulin-like growth factor actions in the central nervous system. Neuropeptides 2002; 36:209-220.
- Geisert Jr EE, Williams RW, Geisert GR, Fan L, Asbury AM, Maecker HT, et al. Increased brain size and glial cell number in CD81-null mice. J Comp Neurol 2002;453:22-32.
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, et al. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature2005; 435:1244-50.
- Haskins A. Macrocephaly Syndromes. Semin Pediatr Neurol. 2007; 14:128-135.
- Ruggieri V, Arberas C. Síndromes genéticos reconocibles en el periodo neonatal. Medicina 2009; 1(69):15-35.
- Phosphatase and tensin homolog; PTEN, Ada Hamosh Online Mendelian Inheritance in Man (OMIM) 2013 OMIM +601728 http://omim.org/entry/601728
- Wynne-Davies R, Walsh WK, Gormley J. Achondroplasia and hypochondroplasia Clinical variation and spinal stenosis. The journal of bone and joint surgery. 1981; 4:509-515.
- Hersh JH. Health Supervision for Children With Neurofibromatosis. Pediatrics 2008; 3:633-642.
- Cecchetto G, Milanese L, Giordano R, Viero A, Suma V, Manara R. Looking at the missing brain: hydranencephaly case series and literature review. Pediatr Neurol. 2013; 48:152-158.
- Iannicelli J, Malla I, Vidoni D, Machi P, Arocena E, Cruz C, et al. Hidrocefalia externa idiopática: una causa de macrocefalia en niños normales. Arch argent pediatr 2002; 100(5): 394-397.
- Castro M, Pérez C, Rodríguez MI, Blanco O, Alonso A, Eirís J. Hidrocefalia externa idiopática benigna (efusión subdural benigna) en 39 niños: evolución natural y relación con la macrocefalia familiar. Rev Neurol 2005; 40(9):513-517.
- Zahl SM, Egge A, Helseth E, Wester K. Benign external hydrocephalus: a review, with emphasis on management. Neurosurg Rev 2011; 34:417–432
- Paciorkowski AR, Greenstein RM. When is enlargement of the subarachnoid spaces not benign? A genetic perspective. Pediatr Neurol 2007; 37:1-7.
- Fransen E, Van Camp G, Vits L, Willems PJ. L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Human Molecular Genetics. 1997; 10:1625-1632.
- Graf WD, Born DE, Sarnat HB.The Pachygyria-Polymicrogyria spectrum of cortical dysplasia in X-linked Hydrocephalus. Eur J Pediatr. 1998; 8:10-14.
- Biran-Gol Y, Malinger G, Cohen H, Davidovitch M, Lev D, Lerman-Sagie T. Developmental outcome of isolated fetal macrocephaly. Ultrasound Obstet Gynecol. 2010; 36:147-153.
- Rubens J, Zepeda G, González A. Klippel-Feil: Una enfermedad musculoesquelética con malformaciones cardiovasculares asociadas. Bol Med Hosp Infant Mex. 2005; 62: 348-355
- Zahl SM, Wester K. Routine measurement of head circumference as a tool for detecting intracranial expansion in infants: What is the gain? A nationwide survey. Pediatrics. 2008; 3(121):416-420.
|
