El enfrentamiento de los pacientes con cuadros infecciosos recurrentes es un problema frecuente en pediatría. Las infecciones pueden darse en varios escenarios generales. Puede tratarse de un paciente normal en un entorno de riesgo, como hacinamiento, vivienda inadecuada, despreocupación de los padres. En otros casos existe un factor individual que las predispone (alteración anatómica, enfermedad crónica, atopia e inmunodeficiencia).
Factores de riesgo de infecciones en niños
- El niño nace con un sistema inmune inmaduro: los recién nacidos poseen inmunoglobulina G (IgG) materna en circulación, la que disminuye en el primer trimestre de la vida mientras se inicia la producción propia, alcanzándose los niveles del adulto, dependiendo del subtipo, alrededor de los dos años de edad. Como no tiene memoria, se hace menos eficiente la respuesta inmune adaptativa, lo que también redunda en una respuesta celular más lenta y menos efectiva. Además, los niveles de complemento en el recién nacido son bajos. Todo lo anterior favorece las infecciones, y de hecho los niños sanos suelen presentar hasta seis episodios de infección respiratoria alta en el primer año.
- La asistencia a guarderías aumenta el riesgo de enfermarse por mayor probabilidad de contagio.
- Se ha visto que la contaminación intradomiciliaria con humo de tabaco favorece las infecciones, además de contribuir al desarrollo de atopia y asma bronquial. De ese modo, los niños inmunocompetentes pueden elevar la frecuencia de infecciones respiratorias altas sólo en relación a esa condición.
- Atopia: la inflamación crónica simula episodios respiratorios infecciosos, y favorece la adherencia de los patógenos al epitelio inflamado.
- Factores anatómicos, incluyendo diskinesia ciliar.
- Cuerpos extraños.
- Fibrosis quística.
- Reflujo gastroesofágico.
Algunas consideraciones con las inmuno-deficiencias secundarias
Estudios en pacientes con desnutrición muestran la presencia de alteraciones especialmente en la respuesta celular. Otras condiciones se asocian a déficit inmunitario, como la prematuridad extrema, la infección por virus de la inmunodeficiencia humana y el tratamiento inmunosupresor. El déficit de vitamina A favorece las infecciones de los tractos respiratorio y gastrointestinal. Defectos enzimáticos o del zinc (acrodermatitis enterohepática) pueden confundirse con una inmunodeficiencia severa combinada. Los anticonvulsivantes, especialmente la hidantoína, pueden desencadenar defectos a nivel de inmunoglobulinas.
En las enfermedades con pérdidas proteicas como síndrome nefrótico, enteropatía perdedora de proteínas, linfangectasia intestinal y grandes quemaduras, puede producirse hipogammaglobulinemia secundaria de difícil manejo.
Otras enfermedades asociadas a defectos inmunitarios son, en otras, el síndrome de Down, y la asplenia anatómica o funcional.
Mecanismos de defensa del huésped e inmunodeficiencias primarias
El sistema inmune nos protege contra invasión por diferentes gérmenes. Los componentes del sistema inmune incluyen la piel y mucosas, las células fagocíticas (polimorfonucleares neutrófilos y células natural killer), el sistema del complemento y otros factores circulantes, y el sistema inmune adaptativo que comprende linfocitos T y B y sus productos. Cualquier alteración en la relación entre estos mecanismo provoca una incapacidad de defenderse por parte del huésped.
Podemos definir algunos patrones de infección que nos orientan al defecto inmune:
- Infecciones recurrentes sino-pulmonares y gastrointestinales después de los 6 meses de edad, generalmente asociadas a defectos humorales.
- Infecciones por gérmenes oportunistas de inicio precoz (retardo del desarrollo pondoestatural, diarreas) que se asocian a defectos celulares.
- Infecciones cutáneas profundas en defectos de la fagocitosis.
- Infecciones por organismos capsulados en defectos del complemento.
Las inmunodeficiencias primarias son en general el resultado de alteraciones genéticas en el desarrollo o función de los linfocitos, y son raras a excepción del déficit de inmunoglobulina A (IgA), que tiene una prevalencia de 1:300 a 1:700 habitantes. La prevalencia estimada de otras inmunodeficiencias es de 1: 10.000 a 1:200.000 dependiendo del diagnóstico.
En forma global, podemos clasificar las inmunodeficiencias en defectos humorales o del linfocito B; defectos celulares o del linfocito T; defectos del complemento y defectos fagocíticos. Las alteraciones humorales son las más frecuentes (alrededor del 50% de todas las inmunodeficiencias), las celulares corresponden al 20 a 30%, los déficits fagocíticos al 18% y del complemento al 2%.
La historia y el examen físico son pilares del diagnóstico, debiendo descartarse alteraciones anatómicas u otras patologías que condicionen inmunodeficiencia secundaria. La frecuencia, duración, severidad, complicaciones y respuesta al tratamiento antimicrobiano son muy importantes. Infecciones que no responden a tratamientos antibióticos adecuados o neumonías multilobares que llevan a la producción de bronquiectasias, y otitis medias que evolucionan hacia mastoiditis son muy sugerentes de una inmunodeficiencia secundaria. Todos los pacientes con bronquiectasias deberían ser evaluados para descartar una inmunodeficiencia de base, especialmente del tipo humoral.
Otros elementos a considerar (tabla 1) son la edad de inicio. Cuando el inicio es posterior a los 7 meses orienta hacia defecto de inmunoglobulinas, ya que el niño está precozmente protegido por el paso transplacentario de inmunoglobulinas y por la leche materna. En cambio los defectos celulares se manifiestan precozmente y ocasionan infecciones severas que comprometen la vida.
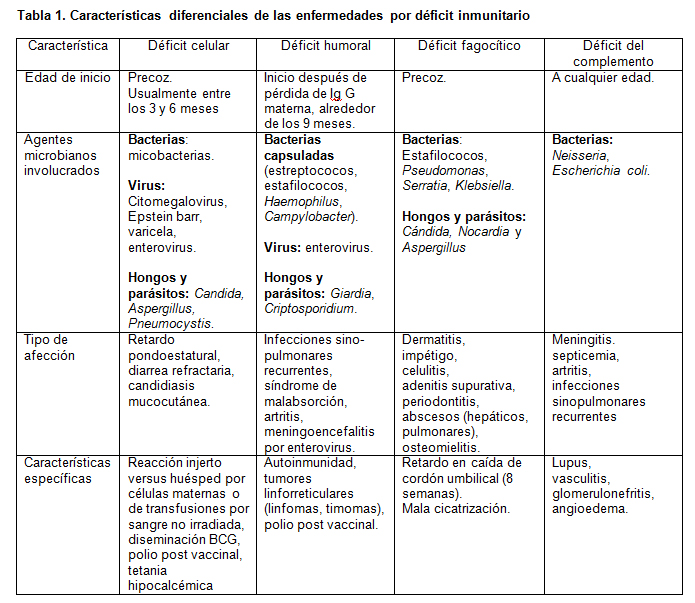
Los gérmenes involucrados son muy importantes: si son gérmenes oportunistas hay que descartar defecto celular desde el inicio. Los linfocitos T son esenciales en el control de hongos, virus, protozoos y bacterias de desarrollo intracelular (micobacterias, Pneumocystis jiroveci, Cándida). Los déficit humorales en general se presentan como infecciones sino-pulmonares por gérmenes capsulados (Streptococcus pneumoniae y Haemophilus influenzae) y meningoencefalitis por enterovirus (coxsackie, echo). A nivel intestinal el déficit de IgA secretora puede presentarse como diarrea crónica secundaria a giardiasis refractaria, infección por Campylobacter, Yersinia o sobrecrecimiento bacteriano intestinal.
La presencia de un absceso hepático por gérmenes catalasa-positivos, sin existir factor predisponente obliga en un niño a descartar enfermedad granulomatosa crónica (alteración de la producción del peróxido de hidrógeno). Otras formas de presentación son adenitis supurada, piodermia y gingivitis recurrente. El retardo de la caída del cordón umbilical después de las 8 semanas o la falta de cicatrización se asocian a defectos de moléculas de adhesión. El síndrome hiper-IgE y de Job se caracterizan por presentar piodermia y neumonías con tendencia a la formación de neumatoceles. Las infecciones recurrentes por Neisseria hacen sospechar defecto en el complejo de ataque de membrana del complemento (C5 a C9). Los déficits de C3 se manifiestan por septicemia, especialmente por bacterias gramnegativas, lo que enfatiza el rol del complemento en la opsonofagocitosis. En cambio los defectos de C2 o C4 se asocian a enfermedades autoinmunes.
El examen físico está orientado a descartar alteraciones sugerentes de inmunodeficiencias secundarias y un examen normal no descarta una inmunodeficiencia primaria. En pacientes con inmunodeficiencias celulares es importante la apariencia: suelen verse gravemente enfermos y
con alteración de su crecimiento pondoestatural, y pueden presentar alteraciones cutáneas sugerentes de reacción de injerto versus huésped.
En la agammaglobulinemia no existe desarrollo de tejido linfoide por lo tanto puede existir ausencia de amígdalas y ganglios.
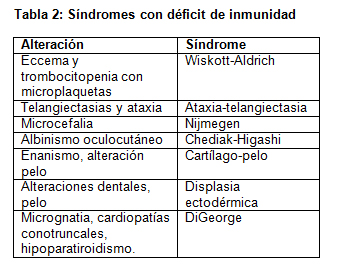
Existe una serie de síndromes con características propias asociadas a inmunodeficiencias (Tabla 2).
Referencias
- Casanova J.-L., Fieschi C., Bustamante J., Reichenbach J., Remus N., von Bernuth H. et al. From idiopathic infectious diseases to novel primary immunodeficiencies. J Allergy Clin Immunol 2005; 116: 426-430.
- Bonilla F., Bernstein I., Khan D., Ballas Z., Chinen J., Frank M. et al. Practice Parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 2005; 94 (suppl): S1-63.
- Notarangelo L., Casanova J., Fischer A., Puck J., Rosen F., Seger R. et al. Primary immunodeficiency diseases: an update. J Allergy Clin Immunol 2004; 114: 677-687.
- Conley M., Stiehm E. Immunodeficiency disorders: General considerations. In Stiehm ER (ed): Immunologic Disorders in Infants and Children. Philadelphia, WB Saunders, 1996, pp 201-252.
- Woroniecka M., Ballow M. Office evaluation of children with recurrent infection. Pediatr Clin North Am. 2000; 47 (6): 1211-24.
- Rich R. Clinical Immunology: Principles and Practice. Mosby- year book. 1996.