Introducción
La enfermedad de Von Willebrand (EvW) es el trastorno hereditario más frecuente de la hemostasia. Fue descrita inicialmente en 1924 por Erich von Willebrand, en Finlandia. Su prevalencia varía de 0,8% a 1%, tanto en la población chilena como extranjera (1). Nilsson en Suecia reportó 1 caso por millón de personas, Rodegheiro y colaboradores en Italia encontraron una prevalencia de 0,82 % y estudios más recientes confirman una prevalencia entre 1 a 2%. (2,3).
La forma habitual de transmisión es autosómica dominante, de alta penetrancia, pero de expresión muy variable inter e intra familiar. Se ha postulado que en algunos casos se hereda como un rasgo autosómico recesivo. Su patogenia está determinada por un defecto cuantitativo o cualitativo del factor von Willebrand (FvW). Debido a que el FvW es también transportador del Factor VIII en plasma, la deficiencia del FvW conlleva a una alteración, tanto en la fase primaria como secundaria de la hemostasia. Las glicoproteínas de alto peso molecular del FvW juegan un rol importante en la fase primaria, ya que favorece la adhesión de la plaqueta al subendotelio y la agregación plaquetaria.
La EvW se debe sospechar en aquel paciente con historia de sangramiento mucocutáneo, sangramiento post operatorio y especialmente en aquellos con una historia familiar sugerente. Los síntomas más comunes son epistaxis, sangramiento post extracción dental y menorragia. La tendencia hemorragípara es altamente variable y depende del tipo y severidad de la enfermedad (3, 4)
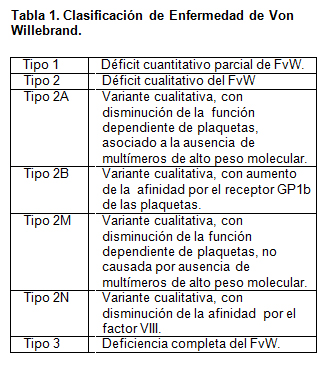
La clasificación se basa en su fisiopatología, diferenciando tres categorías que se detallan en la tabla 1: una deficiencia cuantitativa parcial del FvW en el tipo 1 (clásica) y un defecto cualitativo del FvW en el tipo 2 (variante) y una deficiencia completa del FvW en el tipo 3 (severa). En 1994, Sadler recomienda la subdivisión de la EvW tipo 2, en cuatro variantes 2A, 2B, 2M y 2N, de acuerdo a los detalles específicos de los hechos fenotípicos (4).
Debido a la gran variabilidad clínica y la poca especificidad de los síntomas, el diagnóstico también se debe basar en exámenes de laboratorios, los cuales incluyen: FvW antígeno (Ag), cofactor ristocetina, factor VIII coagulante, análisis de los multímeros del FvW y la capacidad de unión del factor VIII. La relación entre cofactor ristocetina y FvW Ag (Co Ris/Ag) parece ser de utilidad para diferenciar entre enfermedad tipo 1 y tipo 2.
Una correcta identificación de los diferentes tipos y subtipos es importante desde el punto de vista terapéutico, ya que, dependiendo de esto, pueden usarse alternativas de tratamiento y evitar los hemoderivados, dado el riesgo que implican las transfusiones (4, 5, 6, 7)
Los dos tratamientos de elección son: la desmopresina y la terapia transfusional con concentrados de factor VIII/FvW derivados de plasma. Otras alternativas de tratamiento son los antifibrinolíticos y estrógenos. Concentrados de FvW recombinante se han elaborado, sin embargo, sólo han sido probados en animales.
La desmopresina (1-deamino-8–D arginina vasopresina) es un análogo sintético de la vasopresina. Aumenta el factor VIII y FvW sin importantes efectos colaterales. Las ventajas de su uso es el bajo costo y no tiene los riesgos de transmisión viral. Es usada endovenosa, en dosis de 0,3 ug/kg., diluido en 50 ml salina, a pasar en 30 minutos. Este tratamiento aumenta los niveles basales de factor VIII y FvW, 3 a 5 veces dentro de 30 minutos. Las altas concentraciones se mantienen al menos por 6 a 8 hrs. Debido a que la respuesta en un paciente siempre es la misma, una dosis de prueba sirve de ayuda al momento del diagnóstico para identificar su respuesta. Las infusiones pueden ser repetidas cada 12 a 24 horas, dependiendo del tipo y severidad del episodio. La desmopresina es más efectiva en pacientes con tipo 1, especialmente aquellos que tienen depósitos normales de FvW. En otros subtipos la respuesta es variable; en el tipo 2A los niveles de factor VIII se elevan por la acción de la desmopresina (DDAVP), pero el tiempo de sangría se acorta sólo en una minoría de los casos. La desmopresina está contraindicada en el tipo 2B, debido al riesgo de trombocitopenia transitoria (5).
En el tipo 2N un aumento en el nivel de factor VIII se observa después de la administración de la desmopresina, pero por corto tiempo en el plasma, debido a que el efecto estabilizador del FvW está alterado. Los pacientes con tipo 3 usualmente no responden a la desmopresina, sin embargo, un subgrupo de estos pacientes ha sido recientemente identificado en quienes el factor VIII llega a ser normal después de la desmopresina, aunque el tiempo de sangría permanece prolongado (5).
Los aminoácidos antifibrinolíticos son drogas sintéticas que interfieren con la lisis del coágulo por saturación de los sitios de unión del plasminógeno. El ácido tranexámico es el más conocido; puede ser usado oral o endovenoso en dosis de 30 - 50 mg /Kg. / día dividido c / 8 hrs., y también en forma tópica. Se utiliza como coadyuvante en el manejo de sangramiento de cavidad oral, epistaxis, sangramiento gastrointestinal y menorragia en todos los tipos de EvW. Los estrógenos aumentan los niveles plasmáticos de FvW, pero su respuesta es variable e impredecible; son utilizados en reducir la severidad de la menorragia en mujeres con EvW.
Objetivo
Reevaluar un grupo de pacientes pediátricos con diagnóstico de enfermedad de von Willebrand controlados en el Centro Hemofílico del Hospital Roberto del Río, con el fin de precisar el tipo de la enfermedad y relacionar la clínica con los hallazgos de laboratorio.
Material y Método
Un total de 58 pacientes inscritos en el Centro Hemofílico con diagnóstico de EvW aceptaron someterse a una encuesta y nuevo estudio. A todos se les practicó una entrevista protocolizada y se revisaron los antecedentes de su ficha clínica.
La encuesta fue realizada por una sola persona y contestada por el paciente y/o sus padres; ésta consistió en averiguar antecedentes personales y familiares de hemorragia, edad de comienzo y tipo de síntomas, frecuencia de los episodios y terapia transfusional utilizada.
Se les sometió a nuevos estudios de laboratorio en la Unidad Docente asociada de Laboratorio Clínico y Departamento de Hematología de la Universidad Católica de Chile. Estos exámenes consistieron en:
-Tiempo de sangría Ivy (hasta 7 minutos)
-Recuento de plaquetas (150.000 a 450.000 x mm3)
-Actividad del factor VIII: C. (50 – 150 %)
-Actividad cofactor ristocetina. (80 – 150 %)
-Concentración de factor v. W. antigénico en el plasma (0.6 – 1.3 U/ml)
-Estructura multimérica del factor v. W. antigénico.
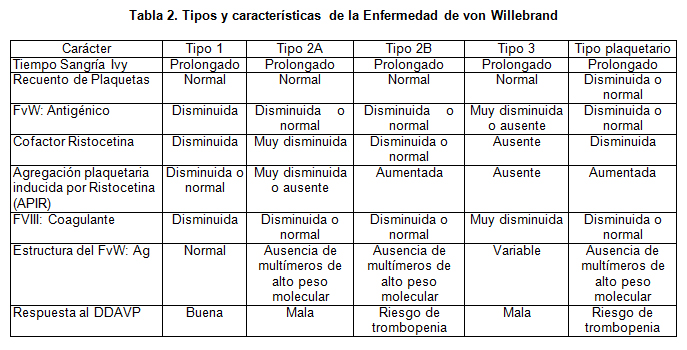
De acuerdo a la encuesta clínica y los exámenes de laboratorio, los pacientes fueron clasificados de acuerdo a la tabla 2 (Tabla modificada de Zimmerman y Ruggeri) (6).
Resultados
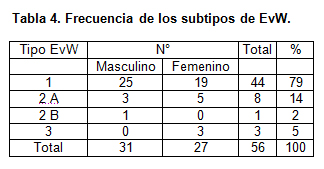
Se estudiaron 58 pacientes portadores de la EvW menores de 15 años, de los cuales 31 fueron de sexo masculino.
La edad de comienzo de las manifestaciones hemorrágicas fue más precoz en relación a la severidad de la enfermedad, con una mediana de consulta a los 4 años en el tipo 1; 2 años en el tipo 2 y menor a 2 años en el tipo 3.
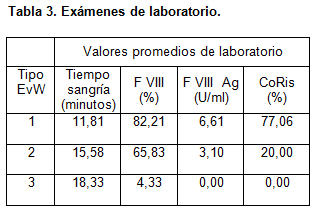
La tabla 3 muestra los valores promedios de los resultados de laboratorio obtenidos de los pacientes en estudio.
De acuerdo a los exámenes realizados, y basado en la clasificación de Zimmerman, de los 58 pacientes, 56 fueron EvW y 2 fueron hemofilia A leve. El 79% correspondió a EvW tipo 1, 16 % tipo 2 y 5 % tipo 3 (tabla 4). El antecedente familiar de sangramiento se encontró en la anamnesis en 78.5 % de los casos.
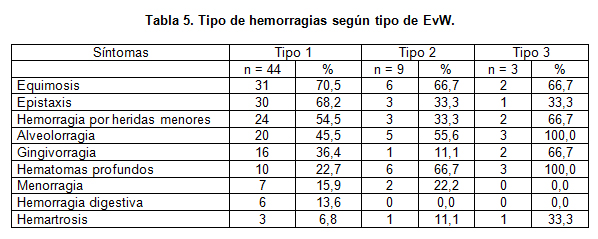
En cuanto a los síntomas, en el tipo 1 y tipo 2 fueron frecuentes las manifestaciones hemorragíparas mucocutáneas clásicas y en cambio en el tipo 3 aparecen hematomas profundos y hemartrosis (tabla 5).
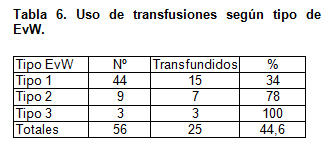
La terapia transfusional utilizada se presenta en la tabla 6, destacando el alto porcentaje de pacientes transfundidos (44,6 %). El 34 % de los pacientes con tipo 1 requirió terapia transfusional.
Discusión
La anamnesis sigue siendo el pilar fundamental en la orientación diagnóstica de las enfermedades congénitas de la coagulación, incluida la EvW. En nuestro trabajo, el 78,5 % de los pacientes tuvieron antecedentes familiares de hemorragias mucocutáneas, frecuencia que aumenta en relación a la mayor severidad de la enfermedad.
Teníamos la impresión que la mayoría de nuestros pacientes eran mujeres, sin embargo, fueron más frecuentes los hombres, tanto en el tipo 1 y 2. El mayor número de consultas corresponden a las mujeres después de la menarquia por menometrorragia; esto no se ve reflejado en nuestros resultados porque la mayoría del grupo estudiado corresponde a pacientes prepúberes.
El diagnóstico de las formas graves de la EvW es relativamente fácil, sin embargo, en las formas leves existe una gran variabilidad en la expresión de laboratorio de la enfermedad en el mismo paciente a lo largo del tiempo, por lo que a este grupo se les debe repetir los exámenes antes de asegurar el diagnóstico. Esta variabilidad explicaría el hecho que dos pacientes inicialmente diagnosticados como EvW resultaron ser hemofílicos A leve.
En esta casuística el tipo 1 fue el más frecuente (79%) y el porcentaje de distribución de los diferentes tipos fue también coincidente con lo encontrado en la literatura nacional y extranjera (4).
Los tipos 2 y 3 son menos frecuentes, pero tienen hemorragias de mayor intensidad y con más frecuencia, por lo tanto la consulta es más precoz y repetida.
Los síntomas más frecuentes observados en nuestros pacientes fueron las hemorragias mucocutáneas, siendo las equimosis, epistaxis y hemorragias secundarias a heridas menores las más frecuentes en los tres tipos clínicos. La frecuencia y severidad de la hemorragia fue variable, pero se relaciona en forma directa con la mayor severidad de la enfermedad (tipo 2 y 3). Los pacientes con EvW tipo 3 tuvieron una severa tendencia hemorrágica, con consultas frecuentes por hemorragia de mucosas y aparición de hematomas profundos y hemartrosis, lo que se relaciona con los bajos niveles de FVIII coagulante encontrado.
El estudio de la composición multimérica del FvW antigénico permite una correcta clasificación, diagnóstico y tratamiento de la enfermedad (8, 9, 10)
En nuestro estudio todos los pacientes con el tipo 3 fueron transfundidos; este porcentaje baja a un 77 % en el tipo 2 y en el tipo 1 sólo un 34 % de los pacientes necesitó en algún momento transfusión con hemoderivados. Si previamente se hubiese conocido el tipo de EvW, este porcentaje quizás sería menor porque precisamente este grupo es el que más se beneficia con alternativas de terapia como es el uso de desmopresina y antifibrinolíticos.
Conclusiones
La correlación clínico-laboratorio demuestra la gran variabilidad de esta patología, por lo cual es necesaria una completa evaluación de laboratorio que incluya el análisis de la composición multimérica del FvW con el fin de catalogar los tipos y subtipos de la enfermedad.
Este estudio es complejo y costoso, por ello se recomienda realizarlo una vez hecho el diagnóstico seguro de EvW, o frente a sospechas fundadas de la enfermedad.
La individualización de los subtipos es imprescindible para un correcto manejo terapéutico, el que depende del tipo de la EvW diagnosticada (11, 12). Así, el objetivo es tratar de identificar los pacientes con tipo 1 de la enfermedad o aquellos que son respondedores a desmopresina, ya que en éstos se evitaría el uso de hemoderivados. Como la acción de la desmopresina es aumentar los niveles circulantes del complejo FvW/FVIII coagulante propio del paciente, por liberación del factor FvW desde el subendotelio, este fármaco es útil en aquellos portadores de la enfermedad con configuración multimérica normal y alteración sólo cuantitativa de los multímeros (tipo 1). En la mayoría de estos pacientes la terapia de elección es la desmopresina y/o antifibrinolíticos, siendo esta modalidad de tratamiento suficiente para controlar la hemorragia y así evitar la transfusión de hemoderivados con todos los riesgos que ella implica. En el tipo 2A de la enfermedad, los niveles de factor VIII generalmente aumentan con la desmopresina, pero el tiempo de sangría sólo se acorta en una minoría de los casos. La desmopresina está contraindicada en el tipo 2B debido a que puede provocar trombocitopenia transitoria. En tipo 2N niveles relativamente alto de factor VIII son observados después de la administración de desmopresina, pero la circulación es de corto tiempo. Los pacientes con tipo III generalmente no responden a la desmopresina (13, 14).
Referencias
- Mezzano D., Pereira J., Quiroga T. Enfermedad de Von Willebrand. Rev. Médica Chile 1990; 118: 320-9.
- Miller Ch., Lenz R., Breen C. Prevalence of von Willebrand’s disease among US adults. Blood 1987; 70: 377-383.
- Werner E., Broxson E., Tucker E., et al. Prevalence of von Willebrand’s disease in children: A multiethnic study. Blood 1991; 78: 68a.
- Sadler J. A revised classification of von Willebrand disease. Thromb Haemost 1994; 71: 520-525.
- Soto V., Morales M., Verdugo P. Empleo del DDAVP en el manejo de Enfermedad de Von Willebrand. Rev Chil Pediatr 2005; 76 (2); 193-197. [Citada 19 julio 2005] Disponible en internet: http://www.scielo.cl/scielo.php?pid=S0370-41062005000200011&script=sci_arttext&tlng=es
- Ruggeri Z., Manucci P., Lombardi R., Federici A., Zimmerman T. Multimeric composition of factor VIII/Von Willebrand factor following administration of DDAVP: implications for pathophysiology and therapy of Von Willebrand`s disease subtypes. Blood 1982; 59: 1272-8.
- Miller J., Castella A. Platelet- type Von Willebrand´s disease: characterization of a new bleeding disorder. Blood 1982; 60: 790-4.
- Quiroga T., Pérez M., Pereira J., Mezzano D. Diagnóstico de los subtipos de Enfermedad de Von Willebrand, mediante análisis de la composición multimerica del factor Von Willebrand plasmático. Rev. Médica Chile 1993, 121: 987-993.
- Mezzano D., Aranda E., Grebe G., Legues M., Rodríguez S., Marzouka M., Ríos E. Diagnóstico de laboratorio de la enfermedad de Von Willebrand en 52 pacientes. Rev. Médica Chile 1983; 111: 1139-44.
- Mezzano D., Grebe G., Vargas L., Lira P., Pereira J. et al. Enfermedad de Von Willebrand. Estudio de 58 pacientes. Rev. Médica Chile 1986; 114:1029-34.
- Zimmermann T., Ruggeri Z. Von Willebrand factor and Von Willebrand disease. Blood 1987; 70: 895-902.
- Ruggeri Z. Structure of Von Willebrand factor and its function in platelet adhesion and thrombus formation. Clin. Hematol 2001; 14: 257-219.
- Sadler J., Mannucci P., Bertorp E., et al. Impact, diagnostics and treatment of Von Willebrand disease throms. Haemost 2000, 84: 160-174.
- Federici A., Mannucci P. Advances in the genetic and treatment of Von Willebrand disease. Curr Op Ped 2002, 14; 23-33