Las crisis epilépticas neonatales ocurren con mucho mayor frecuencia que en cualquier otro período de la vida, teniendo una incidencia variable entre el 0,15 al 1,4% de los recién nacidos vivos, cifra que en los prematuros de 36 semanas o menos aumenta a cerca del 6% (1, 2)
Lanska y colegas han encontrado una incidencia que varía inversamente con el peso de nacimiento. Así, en los menores de 1.500 grs. es de 57,5/1000 RN vivos, 4,4/1000 RN para aquellos con peso entre 1500-2499 grs., 2,8/1000 para los con peso entre 2500 y 3999 grs. y 2/1000 RN vivos para aquellos con más de 4000 grs. (3)
Históricamente se han considerado como crisis a la actividad motora estereotipada, conductas atípicas o anárquicas, o fenómenos secundarios a la activación del sistema nervioso autónomo.
Definición
El término crisis epiléptica define a cualquier evento clínico paroxístico en el cual se sospeche o se haya comprobado la relación con una crisis electroencefalográfica, habitualmente registrada con electrodos colocados en el cuero cabelludo. En otras palabras corresponde a una descarga paroxística, hipersincrónica de un grupo de neuronas corticales. Entre éstas estarían incluidas las crisis clónicas focales y multifocales, las tónicas focales y asimétricas y algunas mioclónicas (4)
Esta denominación no es apropiada para los eventos en los cuales no hay una concomitancia con las crisis electroencefalográficas, en las cuales puede haber involucrado un mecanismo no epiléptico en su origen y propagación. Acorde a Mizrahi y Kellaway (5), en este grupo se pueden incluir las posturas tónicas simétricas generalizadas, algunas mioclónicas y automatismos motores de progresión, movimientos oculares y algunos oro-buco-linguales.
Fisiopatología
Hay conceptos básicos que no han variado en el tiempo, y entre estos, el que la actividad focal clónica es consecuencia de la descarga hipersincrónica de un grupo finito de neuronas corticales. Esta puede permanecer confinada a un sitio altamente específico o esparcirse rápidamente a otras áreas. A su vez, el sistema límbico y las conexiones diencefálicas están más desarrolladas en el niño que en el adulto, lo que explica que estas crisis neonatales se manifiesten frecuentemente como movimientos oculares, cambios en la coloración de la piel especialmente la cara, movimientos bucolinguales, midriasis, apneas.
La despolarización, que implica la rápida introducción del sodio extracelular al interior de la neurona, puede ser afectada por diferentes mecanismos (6), resultando en un fenómeno excesivo. Entre las etiologías encontramos:
- Alteraciones en la producción de energía, que pueden resultar en falla de la bomba de sodio-potasio ATP dependiente, como consecuencia de hipoxemia, isquemia, e hipoglicemia.
- Alteraciones de la membrana celular, que pueden producir cambios en la permeabilidad al sodio. Esto ocurre cuando el calcio y el magnesio interactúan sobre la membrana neuronal, causando la inhibición de los movimientos del sodio. De este modo la hipocalcemia o hipomagnesemia pueden conducir a un aumento en la entrada de sodio y un exceso en la salida de potasio, resultando en una excesiva despolarización.
- Probablemente, una relación anormal entre los neurotransmisores excitatorios e inhibitorios conduzca a una excesiva despolarización, como ocurriría en la dependencia de piridoxina y también en los fenómenos hipóxico-isquémicos.
Probablemente la cantidad y sensibilidad de los receptores de neurotransmisores, especialmente para neurotransmisores excitatorios, tiene gran influencia en producir una despolarización anormal.
Clasificación
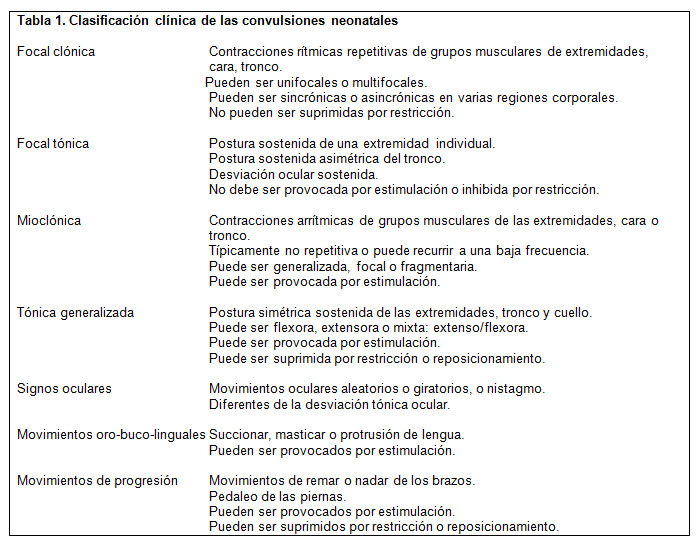
La clasificación clínica (Tabla 1) se ha desarrollado en base a una observación cuidadosa de los fenómenos clínicos observados en los recién nacidos (5), siendo la primera en utilizarse ampliamente.
En esa tabla hay que considerar que los tres últimos grupos de trastornos clínicos son las llamadas crisis neonatales sutiles.
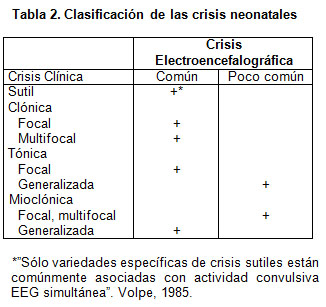
A medida que estas observaciones clínicas se complementaron con EEG, y más recientemente con monitoreo poligráfico-video-EEG, se ha logrado precisar con mayores detalles esta consideración clínica (7, 8) para la cual Volpe (9, 10) ha propuesto la clasificación de la tabla 2.
Otro sistema de clasificación ha sido propuesto por Mizrahi y Kellaway (11), desarrollado a partir de sus hallazgos en los estudios de monitoreo con video-EEG y la presencia o ausencia de actividad epileptiforme electroencefalográfica (Tabla 3).
Cada esquema clasificatorio tiene algunas limitaciones, por lo cual hay una constante revisión y actualización acorde a los avances en el campo. Con relación al período postictal clínico, a menudo hay un mínimo (si es que hay) estado postictal, que sigue posterior a una crisis neonatal focal o localizada. El estado del recién nacido es habitualmente el previo a la crisis, o interictal. Entonces en aquellos que estaban alerta siguen estando alerta, o los que estaban letárgicos o comatosos no modificarán su condición. Alternativamente, aquellos que estaban durmiendo pudieran ser despertados por la crisis. Sin embargo, en aquellas crisis prolongadas o extensas, los neonatos pueden presentar una disminución transitoria en la respuesta a los estímulos.
Actividad eléctrica cerebral
Interictal
A diferencia de lo que ocurre en los niños mayores y en los adultos, en los neonatos no hay marcadores EEG confiables de una epileptogénesis potencial. La actividad de puntas lentas focales puede estar presente en el neonato como una característica del desarrollo normal, o como un hallazgo anormal, sin parecer necesariamente descargas epileptiformes interictales (12).
El análisis del trazado de base del EEG puede ser útil en la evaluación, ya que puede aportar una medida objetiva del grado y severidad de la disfunción neurológica. Así, los EEGs seriados, con el registro inicial obtenido en el período agudo, son los más exactos en caracterizar la evolución del daño cerebral y aportar con información pronóstica. (13, 14)
Existen además anormalidades EEG continuas o periódicas, que pueden tener significado diagnóstico o etiológico específico. Entre ellas están: la actividad estallido-supresión, asociada con la epilepsia precoz mioclónica (incluyendo el síndrome de Ohtahara); el patrón de paroxismos de ritmos theta predominantemente en regiones rolándicas (“theta pointu alternant”), asociado con las convulsiones neonatales benignas (15) el patrón cuasi-periódico focal o multifocal, en encefalitis neonatal por herpes simples (16); y los complejos periódicos de la encefalopatía por glicina (17).
Ictal
La actividad eléctrica crítica es comúnmente focal y bien localizada a una región cerebral relativamente circunscrita, excepto en aquellas descargas asociadas con algunas muecas mioclónicas y con espasmos, que son generalizados. A menudo surgen en el área centrotemporal de un hemisferio y el siguiente sitio en frecuencia es la región occipital. Descargas de la región frontal son relativamente raras (quizás porque se desarrollan más tardíamente). También pueden surgir desde la región medial (Cz) y ser tan confinadas que no se registran en otros sitios.
En un neonato esta actividad eléctrica puede ser unifocal o multifocal. También puede ser simultánea pero independiente, en diferentes regiones cerebrales.
La morfología, frecuencia, y voltaje de la actividad eléctrica varía enormemente en una sola crisis, en tiempos diferentes y en regiones diferentes, y de una crisis a la siguiente. La frecuencia predominante puede estar en el rango de actividad alfa, theta o delta. El voltaje puede ser extremadamente bajo o muy alto. Las descargas en sí mismas pueden ser muy agudas o polimorfas en su configuración, así como sinusoidales. Algunas descargas, ya iniciadas, pueden cambiar gradualmente en voltaje, frecuencia y morfología.
En otras ocasiones comienza y termina abruptamente, virtualmente sin cambios en el carácter (12).
Hay además otros dos patrones eléctricos de crisis que pueden ocurrir en los neonatos, que ocurren típicamente con encefalopatías severas:
-
En el cerebro comprometido, la actividad crítica eléctrica es de bajo voltaje y larga duración, altamente localizada, y muestra poca tendencia a difundir o evolucionar. Puede surgir independientemente de múltiples sitios y puede no ser acompañada por signos clínicos, involucrando habitualmente un mal pronóstico.
-
En el otro patrón, las descargas críticas están en el rango alfa, son sostenidas y rítmicas, 8 a 12 Hz, 20 a 70 mV en la región centro-temporal de un lado. Ellas se han descrito en neonatos con encefalopatía difusa de etiología diversa, y también en algunos con malformaciones cerebrales. (18, 19)
Postictal
Probablemente no existe un estado postictal con características típicas en el EEG. Este puede revertir a su actividad de base previa, sin enlentecimiento postictal como se observa en niños mayores. Sin embargo hay circunstancias, cuando las crisis son frecuentes y prolongadas, en las cuales la actividad EEG postictal se altera, variando de enlentecimiento transitorio a depresión de voltaje, y enlentecimiento a tal grado que el significado clínico no está determinado.
Manejo
El reconocimiento de las crisis clínicas debe iniciar una secuencia de acciones dirigida a confirmar el diagnóstico, evaluación de laboratorio, aproximación etiológica y determinación del pronóstico.
1.-Reconocimiento clínico
El episodio clínico debe ser descrito lo más exacto posible, según lo observado. En base a esto, los fenómenos clínicos pueden clasificarse, y realizarse una determinación de la fisiopatología involucrada. En una circunstancia aguda, esto significará hospitalización y manejo acorde a la condición.
2.-Estudio EEG
El EEG es esencial en la sospecha de crisis neonatales. Debido a que descargas interictales no son absolutamente concluyentes en este grupo etario, lo único que confirma es la presencia de crisis con registro EEG alterado durante el episodio, por lo cual el registro “agudo o crítico” es lo ideal. En algunas circunstancias el registro poligráfico-EEG prolongado es útil, o la detección automática de la actividad crítica EEG, y en otros centros se ha utilizado el monitoreo con video-EEG-poligrafía.
3.-Etiología
Su estudio es esencial, ya que en el neonato las crisis epilépticas pueden ser debidas a una etiología potencialmente tratable. Así debe realizarse una evaluación organizada y pronta, en los aspectos clínicos y de laboratorio, independiente del tipo de crisis y de la presunta fisiopatología.
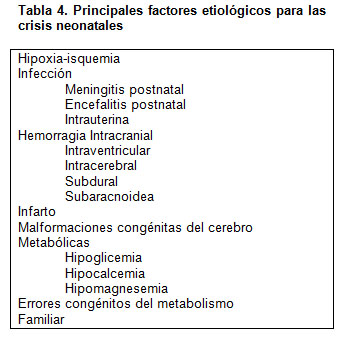
Los principales factores etiológicos se detallan en la tabla 4.
Esta lista señala los principales factores, pero no están en extenso. Ver Goddard et al. (20), y Painter and Gaus (21).
La evaluación de un neonato con crisis debe incluir estudios que de alguna manera estén confirmando o descartando estas etiologías, por lo cual sin duda que requiere: neuroimagen, estudios bioquímicos del líquido cefalorraquídeo, bacteriológicos y virales, entre otros.
Estos factores etiológicos y su importancia, han ido evolucionando acorde ha cambiado la práctica clínica, lo que ha permitido hacer diagnósticos más exactos, por ejemplo en las etiologías infecciosas virales, o en los fenómenos de tipo vascular como los infartos o las hemorragias. También se ha logrado una mayor precisión en las alteraciones anatómicas y el manejo de éstas, así como aquellas relacionadas con trastornos genéticos.
La deficiencia de piridoxina, aunque es citada a menudo, es un hallazgo extremadamente raro.
4.-Pronóstico
El determinante mayor en el pronóstico es la etiología de las convulsiones. Aún no existe consenso en que las convulsiones per se determinen secuelas neurológicas o cognitivas a largo plazo (22).
Entre las secuelas se incluyen el fallecimiento, anormalidades en el examen neurológico, retardo mental y epilepsia postnatal. Las mayores anormalidades ocurren en relación a encefalopatía hipóxico-isquémica, infecciones del sistema nervioso central, y hemorragia cerebral (dependiendo de la extensión y localización). Por otro lado, aquellos con etiología de hipoglicemia e hipocalcemia evolucionaron relativamente normales, en ausencia de otros factores asociados.
Globalmente, impresiona que el determinante más importante de evolución, ya sea fallecimiento o secuela neurológica, es el grado de daño cerebral ocasionado por el agente etiológico (23, 24, 25). Este grado de daño a su vez, puede ser el determinante de otros factores secundarios, tales como el tiempo de inicio de las crisis, tipo de crisis, duración de ellas, patrón EEG ictal e interictal, y dosis y número de fármacos anticonvulsivantes requeridos para el tratamiento.
Los factores de riesgo que aumentan la probabilidad para desarrollar una epilepsia postnatal incluyeron coma y anormalidades significativas de la base del EEG durante el período neonatal, el desarrollo de parálisis cerebral o retardo mental, espigas y puntas lentas, y ondas lentas en los EEG de seguimiento.
Diagnóstico diferencial
Estos episodios paroxísticos deben diferenciarse de las conductas normales observadas en el recién nacido, así como de otros fenómenos paroxísticos.
Muchos fenómenos paroxísticos pueden ser interpretados como anormales por observadores sin experiencia. Así también, otros trastornos pueden considerarse como normales aunque tengan carácter patológico. La Tabla 5 presenta un resumen de los principales, radicando el diagnóstico en las características clínicas de los episodios. En ese contexto es útil diferenciar semiológicamente las convulsiones de los temblores, que se resumen en la tabla 6.


Síndromes específicos que incorporan las crisis como característica esencial
Reconocidos por la Liga Internacional Contra la Epilepsia (International League Against Epilepsy)
La Comisión sobre Clasificación de la ILAE, ha incluido en su Clasificación de Epilepsias y Síndromes Epilépticos (26), síndromes específicos que ocurren en el período neonatal. Las convulsiones neonatales, como una categoría individual de las epilepsias o síndromes epilépticos, están incluidas en la clasificación bajo la categoría (3): Epilepsias y síndromes no determinados que sean focales o generalizados, subtítulos (3.1): con ambas crisis generalizadas y focales.
Otras epilepsias específicas o síndromes con crisis neonatales se listan a continuación:
Convulsiones Neonatales Familiares Benignas
Se presentan al segundo y tercer día de vida (80%). Son crisis clónicas multifocales, a veces con apnea, excepcionalmente tónicas. La duración oscila entre uno a tres minutos, son repetitivas y con una frecuencia de 10 a 20 por día. Pueden presentarse hasta el décimo segundo a decimoquinto día de vida, y aparecer aisladamente durante algunas semanas más. Se piensa que tienen un patrón de transmisión autonómico, basado en el locus en el cromosoma 20 (27), aunque informes recientes sugieren heterogeneidad genética (28). Se considera benigno porque las descripciones clínicas iniciales no informan de secuelas neurológicas en los neonatos afectados. Sin embargo, no todos los niños evolucionan normalmente (29). Se ha clasificado por la ILAE, bajo el título (2): Epilepsias y Síndromes generalizados; subtítulo (2.1): Idiopático, con inicio relacionado a la edad; sin embargo, las crisis clínicas han mostrado ser focales en su carácter (30).
Convulsiones Neonatales Benignas
Suceden en lactantes sin historia familiar de crisis neonatales. Típicamente son recién nacidos de término y con embarazo y parto normal. Las crisis son habitualmente breves, a menudo son clónicas, pueden presentarse en un hemicuerpo o en otro en forma alternante, rara vez son generalizadas, y ocurren entre el cuarto y sexto día de vida. No se logra identificar etiología. Los neonatos son neurológicamente normales antes, durante y después de las crisis (15). Es caracterizado más exactamente como convulsiones neonatales idiopáticas benignas. Debido al período en que se presentan, se referían a ellas inicialmente como las crisis del quinto día (31). También se ha clasificado por la Comisión de clasificación ILAE bajo el título (2): Epilepsias y Síndromes generalizados; subtítulo (2.1): Idiopático, con inicio relacionado a la edad, pero estas crisis también son focales en carácter.
Encefalopatía Mioclónica Temprana o Precoz
Está clasificada por la ILAE bajo la categoría (2): Epilepsias y Síndromes generalizados; subtítulo (2.3): Sintomático; (2.3.1): etiología no específica. Es una constelación de crisis clínicas caracterizadas por la ocurrencia de mioclonus fragmentario, errático, crisis parciales motoras, y espasmos infantiles. Fue descrita en el neonato primero por Aicardi y Goutieres Goutières. El EEG en todos los casos mostraba un patrón de estallido-supresión similar al descrito por Maheshwari y Jeavons (32).
Este patrón electroclínico tiene un significado pronóstico ominoso, y muchos lactantes mueren antes de su primer cumpleaños. La encefalopatía por Glicina es una de las principales causas. Además de la hiperglicinemia no cetótica, hallazgos similares se han reportado en la acidemia D-glicina (33) y en un caso de acidemia propiónica (34).
Encefalopatía Epiléptica Infantil Precoz (Síndrome de Ohtahara)
Ohtahara (35) describió un síndrome caracterizado por espasmos tónicos que ocurrían antes de los 20 días, generalmente en los primeros cinco días, y que carecían de los mioclonos fragmentarios o crisis clónicas descritas por Aicardi y Goutieres Goutières, pero que mostraba el mismo tipo de patrón EEG de estallido supresión. Evolucionan hacia un EEG de tipo hipsarrítmico. Parece probable que sea una variante de la encefalopatía mioclónica precoz. En todos los casos el estado neurológico es grave y evoluciona hacia la muerte o deja secuelas severas. Las causas de este síndrome son múltiples: disgenesias cerebrales, hipoxia isquemia, trastornos congénitos del metabolismo, entre otros.
Tratamiento
El objetivo primario en el manejo de las crisis neonatales es el tratamiento adecuado de los factores etiológicos subyacentes. Consecuentemente, las crisis convulsivas son tratadas con fármacos antiepilépticos y con terapia específica para la etiología.
Sin embargo, estas metas en muchas ocasiones no se logran, ya sea porque no se identifican las etiologías o porque no hay tratamiento efectivo o conocido.
Al enfrentarse a una crisis epiléptica neonatal, se deben considerar siempre los fundamentos básicos de tratamiento que involucran una situación de este tipo, asegurando una vía aérea despejada, con respiración adecuada, y circulación estable, junto a corrección y mantención de los factores metabólicos.
Fármacos antiepilépticos (FAE)
Aunque la elección del fármaco de primera línea es actualmente controvertida, hay consenso en la preferencia de algunos fármacos. Los FAE que se han usado tradicionalmente incluyen el Fenobarbital, Fenitoína, Lorazepam y Diazepam (36, 37).
Fenobarbital
Es el más usado como FAE inicial. La selección del FAE de segunda y tercera línea varía según los centros (38). Hay informes de la eficacia del Fenobarbital para controlar las crisis neonatales después de una dosis de carga inicial (15–20 mg/kg) para alcanzar niveles terapéuticos, en que demuestran una respuesta favorable entre 32% y 36% (39). Un informe adicional de Gal y colaboradores (40), utilizando dosis hasta 40 mg/kg, revela una respuesta de 85%.
Entre los esquemas propuestos, se recomienda Fenobarbital: dosis de carga 20 mg/kg; si las crisis persisten, 5 – 10 mg/kg hasta alcanzar un nivel sanguíneo de 30 – 40 ug/mL. La dosis de mantención fluctúa entre los 3 a 8 mg/kg/día.
Fenitoína
Painter y colaboradores informan que no existe diferencia significativa entre la administración de Fenobarbital y Fenitoína para controlar las crisis. Se recomienda actualmente Fosfenitoína, 20 mg/kg; y luego 5 – 10 mg/kg para alcanzar un nivel sérico de 15 – 25 ug/ml (41).
Lorazepam
La dosis recomendada es de 0,05 – 0,1 mg/kg (hasta 0,15 mg/kg en situación aguda), que puede ser repetida.
Diazepam
La dosis recomendada es de 0,25 mg/kg endovenoso o 0,5mg/kg rectal, aunque en los esquemas actuales tiene un lugar secundario.
Se han utilizado además otros FAE para tratar las crisis epilépticas neonatales, cuando han fallado los fármacos de primera y segunda línea, incluyendo entre estos Midazolam, Valproato endovenoso, Lidocaína, y más raramente Primidona, Carbamazepina y Vigabatrina. (Tabla 7).
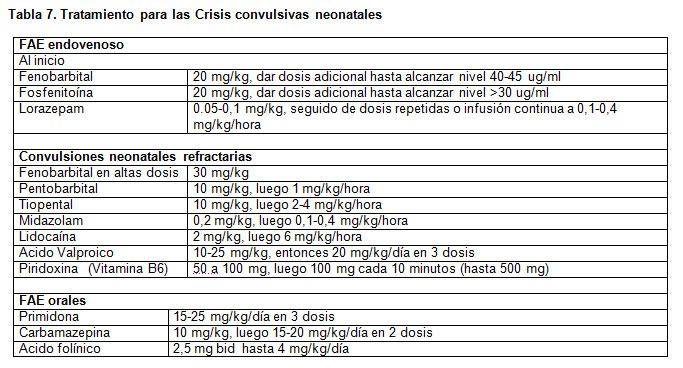
El uso de los FAE se realiza para detener las crisis neonatales, prevenir su recurrencia y minimizar las secuelas que pudieran ocurrir, pero desafortunadamente en muchas ocasiones estos objetivos no se logran.
En múltiples casos la actividad eléctrica epileptiforme puede persistir, planteando la duda de cuál debiera ser el objetivo del tratamiento: cese de las crisis clínicas o de las crisis eléctricas. Aún no hay una respuesta clara de cual debiera ser la meta, y la utilización de altas dosis de FAE para tratar las crisis eléctricas no está exenta de dificultades, como hipotensión sistémica, depresión respiratoria y depresión del sistema nervioso central, entre otras, haciendo la tarea más difícil y a menudo no lograda.
La necesidad de tratamiento crónico con FAE también es materia de controversia, ya que muchos neonatos no tendrán crisis después del período agudo. Habitualmente se les indica tratamiento de mantención con Fenobarbital, y la suspensión de éste es una decisión altamente individualizada, ya que no hay guías prácticas específicas, y la respuesta a los diferentes esquemas no tiene apoyo científico suficiente.
Referencias
- M., Aso K., Beggarly M., et al. Electrographic seizures in pre-term and full-term neonates: clinical correlates, associated brain lesions, and risk for neurologic sequelae. Pediatrics 1993; 91: 128–134.
- Ronen G., Penney S. The epidemiology of clinical neonatal seizures in Newfoundland, Canada: a five-year cohort. Ann Neurol 1995; 38: 518–519.
- Lanska M., Lanska D., Baumann R., et al. A population-based study of neonatal seizures in Fayette County, Kentucky. Neurology 1995; 45: 724–732.
- Mizrahi E. Consensus and controversy in the clinical management of neonatal seizures. Clin Perinatol 1989; 16:485–500.
- Mizrahi E., Plouin P., and Kellaway P. Neonatal Seizures. In Eds. Engel J., and Pedley T. EPILEPSY: The Comprehensive CD-ROM. Lippincot, Williams and Wilkins, 1999.
- Sfaello Z., Bulacio J., Sfaello I., Aicardi J. Crisis epilépticas neonatales. En Campos M.G., Kanner A., eds. Epilepsias. Diagnóstico y tratamiento. Santiago: Mediterráneo, 2004; 185-195.
- Kellaway P., Mizrahi E. Clinical, electroencephalographic, therapeutic, and pathophysiologic studies of neonatal seizures. In: Wasterlain C., Vert P., eds. Neonatal Seizures: Pathophysiology and Pharmacologic Management. New York: Raven Press, 1990; 1–13.
- Mizrahi E., Kellaway P. Characterization and classification of neonatal seizures. Neurology 1987; 37: 1837–1844.
- Volpe J. Neonatal seizures. New Eng J Med 1973; 289: 413–415.
- Volpe J. Neonatal seizures. Pediatrics 1989; 84: 422–428
- Mizrahi E., Kellaway P. Characterization of seizures in neonates and young infants by time-synchronized electroencephalographic-polygraphic-video monitoring. Ann Neurol 1984; 16: 383.
- Hrachovy R., Mizrahi E., Kellaway P. Electroencephalography of the newborn. In: Daly D., Pedley T.A., eds. Current Practice of Clinical Electroencephalography, 2nd ed. New York: Raven Press, 1990; 201–242.
- Tharp B., Scher M., Clancy R. Serial EEGs in normal and abnormal infants with birthweights less than 1200 grams—a prospective study with long term follow-up. Neuropediatrics 1989; 20:.64–72.
- Allan W., Sobel D. Neonatal Intensive Care Neurology. Seminars in Pediatric Neurology, 2004; 11: 119-128.
- Plouin P. Benign neonatal convulsions. In: CG Wasterlain, P Vert, eds. Neonatal Seizures. New York: Raven Press, 1990; 51–59.
- Mizrahi E., Tharp B. A characteristic EEG pattern in neonatal herpes simplex encephalitis. Neurology 1982; 32: 1215–1220.
- Aicardi J., Gotières F. Encephalopathie myoclonique néonatale. Rev Electroencephalogr Neurophysiol Clin 1978; 8: 99–101.
- Dulac O., Aubourg P., Plouin P. Other epileptic syndromes in neonates. In: Rogers J., Dravet C., Bureau M., Dreyfuss F.E., Wolf P., eds. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey, 12–21.
- Willis, G. Periodic alpha seizures with apnea in a newborn. Dev Med Child Neurol 1980; 22: 214–222.
- Goddard J., Glaze D., Fishman M. Neurological disorders of the neonate. Curr Neurol 1982; 4: 241–261.
- Painter M., Gaus L. Neonatal seizures: diagnosis and treatment. J Child Neurol 1991; 6: 101–108.
- Holmes G. Do seizures cause brain damage? Epilepsia 1991; 32(Suppl 5): S14-S28.
- Bergman I., Painter M., Hirsch R., et al. Outcome in neonates with convulsions treated in an intensive care unit. Ann Neurol 1983; 14: 642–647.
- Holden K., Mellits E., Freeman J. Neonatal seizures. I. Correlation of prenatal and perinatal events with outcomes. Pediatrics 1982; 70: 165–176.
- Kellaway P., Mizrahi E. Neonatal seizures. In: H. Luders Lüders, RP Lesser, eds. Epilepsy: Electroclinical Syndromes. New York: Springer-Verlag, 1987; 13–47.
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30: 389–399.
- Leppert M., Anderson V., Quattlebaum T., et al. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nature 1989; 337: 647–648.
- Lewis T., Leach R., Ward K. et al. Genetic heterogeneity in benign familial neonatal convulsions: identification of a new locus on chromosome-8q. Am J Human Genet 1993; 53: 670–675.
- Ronen G., Rosales T., Connolly M. et al. Seizure characteristics in chromosome 20 benign familial neonatal convulsions. Neurology 1993; 43: 1355–1360.
- Hirsch E., Velez A., Sellal F., Maton B., Grinspan A., Malafosse A., Marescaux C. Electroclinical signs of benign neonatal familial convulsions. Ann Neurol 1993; 34: 835–841.
- Dehan M., Quileron D., Navelet Y. et al. Les convulsions du 5e jour. Arch Fr Pediatri 1977; 34: 730–742.
- Maheshwari M., Jeavons P. The prognostic implications of suppression-burst activity in the EEG in infancy. Epilepsia 1975; 16: 127–131.
- Brandt N., Rasmussen K., Brandt S., Schonheyder F. D-glyceric acidemia with hyperglycinemia. A new inborn error of metabolism. Br Med J 1974; 4: 334–336.
- Vigevano F., Bosman C., Giscondi A., Maccagnani F., Sevanti G., Sergo M. Neonatal myoclonic epileptic encephalopathy without hyperglycinemia. Electroencephalogr Clin Neurophysiol 1981; 52: 52P-53P.
- Ohtahara S. Clinico-electrical delineation of the epileptic encephalopathies in childhood. Asian Med J 1978; 21: –17.
- Maytal J., Novak G., King K. Lorazepam in the treatment of refractory neonatal seizures. J Child Neurol 1991; 6: 319–323.
- Painter M., Alvin J. Choice of anticonvulsants in the treatment of neonatal seizures. In: Wasterlain C.G., Vert P., eds. Neonatal Seizures. New York: Raven Press, 1990; 243–256.
- André M., Vert P., Wasterlain C. To treat or not to treat: A survey of current medical practice toward neonatal seizures. In: Wasterlain C., Vert P., eds. Neonatal Seizures. New York: Raven Press, 1990; 303–307.
- Lockman L., Kriel R., Zaske D. Phenobarbital dosage for control of neonatal seizures. Neurology 1979; 29: 1445–1449.
- Gal P., Tobock J., Boer H., Erkan N., Wells T. Efficacy of phenobarbital monotherapy in treatment of neonatal seizures—relationship to blood levels. Neurology 1982; 32: 1401–1404.
- Sfaello Z., Bulacio J., Sfaello I., Aicardi J. Crisis epilépticas neonatales. En Campos M.G., Kanner A.M., eds. Epilepsias. Diagnóstico y tratamiento. Santiago: Mediterráneo, 2004; 185-195.
- Riviello J., Holmes G. The treatment of Status Epilepticus. Seminars in Pediatric Neurology, 11: 129-138, 2004.
|