La niñez es, por definición, una etapa de crecimiento y desarrollo. Muchas consultas en pediatría y neurología pediátrica se relacionan con el retraso en la adquisición de nuevas habilidades, estimándose que los trastornos del desarrollo afectan a alrededor del 10% de la población infantil. Mucho menos frecuentes, pero más dramáticos, son los casos en que existe pérdida progresiva de habilidades. Frente a esta situación, habiéndose descartado tumor o hidrocefalia, deben investigarse causas genéticas, alteraciones metabólicas, infecciones y acción de tóxicos. En ese contexto, las lipofuscinosis ceroides neuronales se consideran el trastorno neurodegenerativo de depósito más frecuente de la infancia (1, 2).
Las lipofuscinosis ceroideas neuronales (LCN, y en inglés NCL) son un grupo de enfermedades neurodegenerativas con herencia autosómica recesiva (3), que se presentan principalmente en la infancia y adolescencia, caracterizadas por síntomatología variable que incluye convulsiones, deterioro cognitivo, pérdida visual y/o atrofia cerebral. El curso es habitualmente progresivo, con desarrollo de demencia que lleva a la muerte (4). Desde el punto de vista neuropatológico se caracterizan por la acumulación progresiva de lipofuscina, un lipopigmento autofluorescente, en neuronas y otros tejidos.
Producidas por mutaciones en distintos genes, estas enfermedades presentan en conjunto una prevalencia de 0,1 a 7 por cada 100.000 recién nacidos vivos (3). En la población finlandesa se ha observado una prevalencia de 1 por cada 12.500 recién nacidos vivos (5).
Historia
La descripción original fue realizada en 1826 por Stengel en Noruega, y la primera sistematización y correlación anatomoclínica la hizo el médico francés Frederick Battenen 1903, por lo que el cuadro llevó inicialmente su nombre. Se consideró en un principio como una variante de la enfermedad reportada por Sachs en 1887. Sin embargo, fue el mismo Batten quien en 1914 concluyó que este cuadro no tenía relación con la enfermedad de Tay-Sachs,sino que correspondía a una entidad clínica nueva (6).
Las lipofuscinosis fueron agrupadas inicialmente bajo el nombre de “idiocia familiar amaurótica”, intentando destacar así sus dos principales características: compromiso intelectual y visual. En el año 1969, Zeman y Dykenacuñaron el término “lipofuscinosis ceroidea neuronal” y luego de múltiples descripciones de otras patologías que compartían características similares, se propuso la primera clasificación realizada por Norman y Wood. Con posterioridad se han planteado otras, a medida que se han identificado los materiales de depósito intracelular.
En 1998, Wisniewski (7)propuso unaclasificación en la que distingue cinco grupos principales, separados de acuerdo a las características clínicas y a la edad de inicio de los síntomas de cada patología. Esta clasificación se ha ido actualizando con el aporte de la genética y de la morfología microscópica (4).
El término lipofuscinosis ceroidea neuronal se ha mantenido, enfatizando la presencia de acumulación de lipofuscina, pese a que no es la causa del daño celular.
Patogenia
La lipofuscina es un polímero intralisosomal compuesto de lipoproteínas residuales de procesos oxidativos. Frecuentemente llamada el “pigmento de la edad”, es considerada el marcador del envejecimiento, ya que aumenta con la edad de un modo casi lineal, observándose frecuentemente en células parenquimatosas de órganos o tejidos con atrofia normal o patológica, neuronas del sistema nervioso central y de ganglios simpáticos, en la zona fascicular de la corteza suprarrenal y en el epitelio de las vesículas seminales (8). Su nombre viene del griego lipo (grasa) y del latín fuscus (oscuro). Los productos de desecho celular se acumulan en autofagosomas. A éstos se unen lisosomas constituyéndose los autofagolisosomas, en los que se realiza la degradación a productos que vuelven a ser utilizados por la célula. Este es un proceso fisiológico donde teóricamente no debería sobrar nada, sin embargo se produce una desviación hacia la peroxidación de lípidos con formación de ácidos grasos insaturados, que se acumulan como residuos visibles al microscopio de luz bajo la forma de gránulos de lipofuscina. La sudanofilia se va perdiendo en estos gránulos a medida que los ácidos grasos no saturados se van transformando, dando origen a este pigmento autofluorescente amarillo-café (8).
En las lipofuscinosis el depósito de material proteico en lisosomas de distintos grupos celulares toma una forma característica según el tipo de LCN estudiada. Pese a que estos cúmulos se producen en distintas células, la muerte celular aparece específicamente en neuronas del sistema nervioso central y de la retina, aparentemente por la inexistencia de actividad mitótica de éstas. Se han obtenido distintos modelos animales (en especial ratas) que presentan las mutaciones descritas para las variantes de LCN. Al estudiar estos modelos se aprecia que la aparición del cuadro clínico es similar entre los distintos modelos y se correlaciona estrechamente con el humano, lo que hace plantear que existe un momento crítico en que se activan los mecanismos de destrucción celular que llevan a la expresión clínica, hipótesis que en la actualidad es objeto de estudio, dado que tiene la potencialidad de convertirse en una ventana terapéutica (9).
Genética
Las lipofuscinosis son cuadros de herencia autosómica recesiva, a excepción del tipo 4 (LCN4), que podría heredarse de forma autosómica dominante (10). En los años 90 fueron identificados cinco genes asociados a estas patologías, como también la secuencia y función de los productos proteicos de los genes para LCN1 y LCN2. Posteriormente se completó la identificación de seis genes (LCN1, LCN2, LCN3, LCN5, LCN6 y LCN8) (11), se identificaron los productos de LCN3, LCN5 y LCN8, y se avanzó en la identificación de al menos 150 mutaciones diferentes en los genes descritos, asociadas a diversos fenotipos (12). Estas mutaciones se encuentran actualizadas en la base de datos MRC Laboratory for Molecular Cell Biology, University College London (www.ucl.ac.uk/ncl/).
A pesar de que la investigación ha aportado nuevos conocimientos en torno a las LCN, queda aún mucho por dilucidar tanto en relación a otros genes y mutaciones implicados, como a sus productos proteicos.
Cuadro clínico
En función de la edad de comienzo, el curso clínico y la morfología ultraestructural, las LCN se han clasificado en cuatro tipos principales:
- LCN infantil (enfermedad de Haltia-Santavuori, LCN1), hallada predominantemente en Finlandia.
- LCN infantil tardía (enfermedad de Jansky-Bielschowsky, LCN2).
- LCN juvenil (enfermedad de Batten, enfermedad de Spielmeyer-Vogt-Sjorgen, LCN3).
- LCN del adulto (enfermedad de Kufs)
Se han descrito además variantes o formas atípicas que representan alrededor de un 20% de las LCN en diferentes poblaciones, generalmente distribuidas entre los grupos infantil tardío y juvenil. Para la forma infantil tardía se han descrito la variante finlandesa (LCN5) (13,14) y la variante LCN6 (15). Existe otra variante conocida como LCN8, epilepsia del norte o epilepsia progresiva con retardo mental (EPRM). Los casos de la forma turca infantil tardía pueden representar variantes de la EPRM. Resumen en Tabla 1.
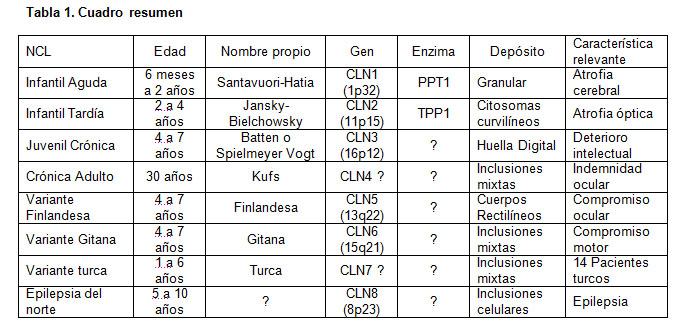
1.LCN1 o lipofuscinosis aguda infantil (Santavuori-Haltia).
Su primera descripción fue realizada por Muldegrer en1903. Sin embargo, fue Santavuori en 1973 quien sistematizó y dio su nombre al cuadro (15).
Se inicia habitualmente entre los 6 meses y los 2 años de vida con aparición relativamente aguda de déficit motor, hipotonía e irritabilidad, a los que siguen crisis convulsivas, generalmente mioclonías, ataxia y finalmente ceguera y grave compromiso cognitivo. Se acompaña de microcefalia adquirida y atrofia cerebral severa con pérdida neuronal en la corteza cerebral y cerebelosa, en la médula espinal y tronco cerebral (4). La mayoría de los pacientes fallece entre los 5 y 10 años.
La enfermedad de Santavuori-Haltia se relaciona con diversas mutaciones en el gen LCN1, en el cromosoma 1p32, de las cuales hay al menos 41 descritas hasta ahora (11) y que determinan el déficit de una enzima lisosomal denominada tioesterasa proteinopalmitoil 1 (o PPT1). El diagnóstico se establece midiendo la actividad de esta enzima en leucocitos y cultivos de fibroblastos, aunque también se puede realizar mediante la genética molecular, identificando las mutaciones del caso índice.
En la resonancia magnética de encéfalo lo más característico es la pérdida de intensidad de señal talámica en secuencias T2, adelgazamiento del cuerpo calloso y aumento de señal periventricular, seguidos de atrofia cerebelar y atrofia cerebral difusa que se estabiliza alrededor de los 4 años. Posteriormente se describe un aumento difuso de señal de sustancia blanca y en la espectroscopía se aprecia pérdida de N-acetil-aspartato y reducción de creatina con aumento de mioinositol y lactato en sustancia gris y blanca. En el Spect (Single Photon Emission Computed Tomography) se describe hipoperfusión cerebelar y cerebral progresiva con conservación de la perfusión de ganglios basales (10).
El estudio enzimático se recomienda como primer análisis frente a la sospecha clínica. Si hay deficiencia de actividad enzimática, se hace estudio molecular. El diagnóstico de aproximación se establece con el análisis histológico de muestra de piel - donde se observan predominantemente depósitos osmofílicos granulares en la microscopía electrónica - y se confirma midiendo la actividad de esta enzima en leucocitos y cultivos de fibroblastos. También puede realizarse mediante genética molecular (10).
2.LCN2 o lipofuscinosis infantil tardía. (Bielschowsky-Jansky).
Conocida como enfermedad de Bielschowsky, su primera descripción fue realizada en 1908 por Jansky.
Se caracteriza por iniciarse entre los 2 y 4 años con crisis tónico-clónicas generalizadas, ausencias o crisis parciales secundariamente generalizadas, aunque su característica típica es la mioclonía, que aparece posteriormente junto a pérdida cognitiva, ataxia, mioclonías, signos extrapiramidales y piramidales, y deterioro rápidamente progresivo de la visión por atrofia óptica. Las crisis convulsivas se asocian a pérdida o falta de adquisición de habilidades del desarrollo psicomotor. El paciente puede fallecer en este primer episodio o derivar a estado vegetativo permanente por años.
Las neuroimágenes muestran atrofia cerebral y cerebelosa progresiva con indemnidad de tálamos y ganglios basales (12).
La tomografía por emisión de positrones (PET) muestra hipometabolismo generalizado de la glucosa (10) y en el análisis histológico se observan depósitos de citosomas curvilíneos, aunque puede haber inclusiones lisosomales mixtas (12).
La lipofuscinosis infantil tardía se debe a diversas mutaciones en el gen LCN2 ubicado en el cromosoma 11p15, que determinan la deficiencia de una enzima lisosomal, la tripeptidil-peptidasa o TPP1. Hasta ahora hay al menos 52 mutaciones descritas (11).
Los estudios neurofisiológicos son importantes para el diagnóstico. El electroencefalograma (EEG) se altera precozmente, presentando una disfunción lenta difusa asociada a actividad epileptiforme multifocal y descargas de punta-onda y poliespiga-onda interictales e ictales. Con la estimulación fótica de baja frecuencia (1 a 2 Hz) se obtiene una respuesta característica de puntas de alto voltaje (mayor a 200 mV) en regiones posteriores, seguidas de una onda lenta que corresponde a un potencial evocado visual gigante El electrorretinograma es usualmente anormal desde el inicio del cuadro con extinción precoz de la respuesta por alteración de conos y bastones (16, 17, 18).
El diagnóstico de LCN2 se puede confirmar a través de la medición de la actividad de la TPP1 en linfocitos o fibroblastos. El diagnóstico prenatal se realiza mediante el análisis de la actividad enzimática en vellosidades coriales o líquido amniótico, o mediante la búsqueda de mutaciones de los casos índice y los portadores (12).
3.LCN3 o lipofuscinosis juvenil crónica (Spielmayer-Vogt-Batten)
Conocida como enfermedad de Batten, en honor al responsable de la primera descripción realizada en 1903, es la más común de las lipofuscinosis en la población norteamericana, correspondiendo al 45 a 52% de los casos. Tiene característicamente dos formas fenotípicas, la clásica (OMIM 204200) y la retardada o tardía (12). En la forma clásica los primeros síntomas aparecen entre los 4 y 7 años de vida, con pérdida progresiva de la capacidad visual, apreciándose degeneración pigmentaria de la retina y atrofia óptica. Posteriormente se hace evidente el deterioro intelectual progresivo, con trastornos del habla, pérdida de las funciones cognitivas y crisis convulsivas tónico-clónicas generalizadas o parciales complejas, raramente mioclónicas (10). En la segunda década de la vida se hacen más prominentes los trastornos de comportamiento, signos extrapiramidales y trastornos del sueño.
En el estudio histológico de tejido rectal, piel y conjuntivas son característicos los linfocitos vacuolados y citosomas en forma de huella digital (10).
Las neuroimágenes pueden mostrar atrofia cerebral y sólo tardíamente cerebelosa. No existe aún una alteración enzimática identificada pero se sabe que el gen LCN3 se encuentra en el cromosoma 16p12 y codifica para una proteína estructural del lisosoma (4), habiéndose descrito hasta ahora 31 mutaciones asociadas a distinta presentación clínica (11).
4.LCN4, lipofuscinosis crónica del adulto (Enfermedad de Kufs).
Descrita por Kufs en 1925, los síntomas iniciales aparecen alrededor de los 30 años, siendo una de sus características relevantes la indemnidad ocular. Clásicamente existen 2 fenotipos: un cuadro caracterizado por epilepsia mioclónica progresiva con demencia, ataxia y signos piramidales y extrapiramidales tardíos, y otro con trastornos del comportamiento y demencia que se pueden asociar a ataxia y signos extrapiramidales (9). En la microscopía electrónica se observan inclusiones mixtas lisosomales.
Su genotipo es desconocido ya que el gen LCN4 aún no se ha tipificado.
Formas atípicas o variantes
1.LCN5, lipofuscinosis infantil tardía (variante finlandesa).
Se manifiesta más tardíamente que la forma infantil tardía (Jansky-Bielchowsky) clásica, entre los 4 y 7 años de edad, con características clínicas muy similares a esta forma, pero a diferencia de ella el compromiso ocular es el síntoma más relevante. En el análisis por microscopía electrónica se observa material de inclusión del tipo cuerpos rectilíneos y en huella digital. El gen, cuyo defecto causa la enfermedad, se encuentra en el cromosoma 13q22 y, aunque ya se han descrito al menos cuatro mutaciones, aún se desconoce la función de su producto (11).
2.LCN6, lipofuscinosis infantil tardía (variante checa o gitana o india).
Esta forma de lipofuscinosis es más frecuente en poblaciones gitanas europeas. El gen asociado, LCN6, está ubicado en el cromosoma 15q21-23, existiendo 18 mutaciones conocidas hasta ahora. Su producto proteico está siendo caracterizado (11).
Respecto al cuadro clínico, predominan los síntomas motores sobre los oculares. En la microscopía electrónica es posible observar inclusiones celulares mixtas: citosomas curvilíneos, en huella digital y cuerpos rectilíneos.
3.LCN7, lipofuscinosis infantil tardía (variante turca).
Descrita el año 1999, se inicia entre el primer y sexto año de vida, con pérdida de las habilidades motrices, deterioro visual y deterioro cognitivo. Se han registrado 14 pacientes, todos turcos y la mayoría con relación de parentesco (19).
El gen LCN7 no ha sido aún mapeado ni su producto proteico definido. En algunos de los pacientes inicialmente descritos se ha observado una mutación alélica de LCN8 y deben, por lo tanto, realizarse estudios para demostrar si esta patología existe como una entidad clínica separada (10). En la histología se observan inclusiones celulares mixtas: citosomas curvilíneos, en huella digital y cuerpos rectilíneos.
4.LCN8, LCN infantil tardía (epilepsia del norte).
Descrita en 1999, se presenta clínicamente con epilepsia tónico-clónica o parcial compleja con retardo mental y disfunción motriz. Los síntomas se presentan entre los 5 y 10 años de vida (20).
El gen LCN8 se encuentra en el cromosoma 8p23 y codifica para una proteína estructural del retículo endoplásmico (21). Se han descrito 5 mutaciones con gran variabilidad fenotípica (7).
El diagnóstico se realiza por medio del análisis histológico, en que se detectan inclusiones celulares o depósitos granulares osmofílicos, y se confirma por medio del análisis de las mutaciones del gen LCN8.
Diagnóstico
El primer pilar del diagnóstico de una lipofuscinosis es el cuadro clínico, que característicamente se presenta como un trastorno rápido o más lentamente progresivo, con alteraciones visuales, epilepsia, demencia y/o alteraciones motrices, con inicio a distintas edades. Alrededor de un 20% de los casos no se enmarca claramente en esta descripción (10).
Existe consenso en la utilidad del estudio por microscopía electrónica de leucocitos o piel, en busca de las inclusiones características, aunque existen falsos negativos. Según la sospecha diagnóstica, deben realizarse dirigidamente los estudios enzimáticos y de genética molecular disponibles.
Dada la variabilidad de las presentaciones clínicas en las distintas poblaciones, debe considerarse la etnia del paciente.
Tratamiento
No existe un tratamiento específico. El manejo se orienta desde las fases iniciales a mejorar la calidad de vida del paciente y su familia con medidas generales y de sostén. Se ha recomendado el aporte de selenio y vitamina E, aunque no existen aún estudios randomizados que permitan demostrar su efectividad (4). El uso de lamotrigina y carbamazepina ha demostrado cierta efectividad para el control de las crisis epilépticas (22).
El trasplante de células madre o troncales (stem cells) ha mostrado una desaceleración leve del deterioro cognitivo y visual en tres casos de la forma aguda infantil. La actividad de la enzima PPT1 se normalizó en leucocitos periféricos, pero se mantuvo en niveles bajos en el LCR. Todos los pacientes que lo recibieron desarrollaron la enfermedad a la edad de 2 o 3 años (23). No hay evidencias sobre la utilidad del trasplante de médula ósea (24).
Otros tratamientos en etapa de investigación, como reemplazo enzimático, terapia génica, administración de cisteamina no han mostrado aún efectos beneficiosos. La cisteamina, aprobada para el uso en cistinosis, también una enfermedad de depósito lisosomal, provoca in vitro disrupción del enlace proteolipídico y reducción de los depósitos lisosomales, y previene la apoptosis en células de pacientes con deficiencia de PPT1 (24).
Durante la última década se han realizado extraordinarios progresos en la tipificación y el conocimiento de los mecanismos patogénicos involucrados en estas enfermedades. La esperanza para el futuro es encontrar un tratamiento que evite la progresión del deterioro neurológico, para lo cual el desarrollo de modelos animales de la enfermedad ha sido crucial. Tan importante como lo anterior se considera el estudio de los pacientes afectados, en relación a la tipificación y curso de la clínica en las diversas formas y al control del grado de progresión. En la medida que se desarrollen terapias específicas para estas enfermedades será esencial realizar el diagnóstico correcto en forma precoz, antes que el daño irreversible haya ocurrido.
Agradecimientos A la Dra. María de los Ángeles Avaria por su contribución, sugerencias y corrección del texto.
Referencias
- Aberg L. Juvenile Neural ceroid lipofuscinosis; Brain-Related symptoms and their treatment. Academic Dissertation, Faculty of Medicine of the University of Helsinki 2001.
- Santavuori P. Neural ceroid lipofuscinoses in Childhood. Brain Dev 1988; 10: 80-83.
- Zeman W., Dyken P. Neural Ceroid Lipofuscinosis: Relationship to amaurotic family idiocy? Pediatrics 1969, 44: 570-583.
- Goebel H, Symposium: The Neural ceroid-lipofuscinoses (NCL). A group of lysosomal diseases come of age. Introduction. Brain Pathol 2004; 14: 59-60
- Vesa J. Hellsten E. Mutations in the palmitoyl protein thioesterase gene causing infantile neural ceroid lipofuscinosis. Nature 1995; 376: 584-587.
- Celia H. Neural ceroid lipofuscinosis. eMedicine February 20, 2002. [Citada Internet 20 abril 2005]. http://www.emedicine.com/neuro/topic498.htm
- Wisniewski K.E. A new classification of neural ceroid lipofuscinoses based on the clinicopathologic and genetic information. Academic Press San Diego 1998; 44: 577
- Brunk U.T., Terman A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function Free Radical Biology & Medicine, Vol. 33, 2002 No. 5, pp. 611–619.
- Mitchison H., Lim M.J. Selectivity and types of cell death in the Neural Ceroid Lipofuscinoses. A group of lysosomal diseases come of age Current state of clinical and morphological features in Human NCL. Brain Pathol 2004; 14: 86-96.
- Goebel H. Wisniewski K.E. Symposium: The Neural ceroid-lipofuscinoses (NCL) A group of lysosomal diseases come of age. Current state of clinical and morphological features inf Human NCL. Brain Pathol 2004; 14: 61-69.
- Mole S. Symposium: The Neural ceroid-lipofuscinoses (NCL). A group of lysosomal diseases come of age. The Genetic Spectrum of Human Neural Ceroid Lipofuscinosis. Brain Pathol 2004; 14: 70-76.
- Wisniewski K.E. Pheno/genotypic correlations of neural ceroid lipofuscinosis. Neurology. 2001; 57(4): 576-81.
- Haltia M. The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol 2003; 62: 1-13.
- Savukoski M., Klockars T., Holmberg V., Santavuori P., Lander E.S., Peltonen L. CLN5 A novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis.Nat Genet 1998; 19: 286-8.
- Santavuori P., Haltia M. Infantile type of so-called neural ceroid-lipofuscinosis. 1. A clinical study of 15 patients. J Neurol Sci 1973; 18: 257-267.
- Aicardi J., Plouin P., Goutières F. Ceroid-lipofuscinosis. Electroencephalogr Clin Neurophysiol 1978; 8: 149-59.
- Pampiglione G., Harden A. So-called neuronal ceroid lipofuscinosis. Neurophysiological studies in 60 children. J Neurol Neurosurg Psychiatry 1977; 40: 323-30.
- Veneselli E., Biancheri R., Buoni S., Fois A. Clinical and EEG findings in 18 cases of late infantile neuronal ceroid lipofuscinosis. Brain Dev 2001; 23: 306-11.
- Wheeler R., Sharp J., Mitchell W., Bate S., Williams, R., Lake, B., Gardiner R. A new locus for variant late neuronal ceroid lipofuscinosis-CLN7. Molec Genet Metab 1999; 66: 337-338.
- Ranta S., Zhang Y., Ross B., Lonka L., Takkunen E., Messer A., Sharp J., Wheeler R., Kusumi K., Mole S., Liu W., Soares M., de Fatima Bonaldo M., Hirvasniemi A., de la Chapelle A., Gilliam T., Lehesjoki A. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nature Genet 1999; 23: 233-236.
- Lonka L., Kyttala A., Ranta S., Jalanko A., Lehesjoki A.-E. The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Hum Molec Genet 2000; 9: 1691-1697.
- Laura A., Juha R. A favorable response to antiparkinsonian treatment in juvenile neural ceroid lipofuscinosis. Neurology 2001 ; (9) : 56.
- Lonnqvist T., Vanhanen S., Vettenranta K., Autti T., Rapola J., Santavuori P., Saarinen-Pihkala U. Hematopoietic stem cell transplantation in infantile neuronal ceroid lipofuscinosis. Neurology 2001 Oct 23; 57 (8): 1411-6.
- Zhang Z., Butler J., Levin S., Wisniewski K., Brooks S., Mukherjee A. Lysosomal ceroid depletion by drugs: therapeutic implications for a hereditary neurodegenerative disease of childhood. Nat Med 2001 (4): 478-84.
|
