La muerte súbita (MS) se define como la muerte abrupta e inesperada en ausencia de anomalías cardiovasculares conocidas (1). Muerte súbita cardíaca (MSC), corresponde a la “muerte rápida e inesperada, de causa cardíaca, que ocurre dentro de la primera hora de iniciado los síntomas, producto de una severa disfunción o cese de la actividad eléctrica y mecánica del corazón, resultando en la pérdida de conciencia y colapso casi instantáneos” (2).
Hasta un 90% de los casos de MS son de causa cardíaca y a pesar de que la MSC puede ser la primera manifestación de patología cardíaca, un gran porcentaje de los casos son potencialmente prevenibles (1). Entre un 25% y 60% de los pacientes con MSC en población pediátrica presentan algún síntoma previamente (síncope, palpitaciones, dolor torácico, disnea) (3). En lo anterior radica la importancia de reconocer síntomas y signos que traduzcan un riesgo cardiológico, para dirigir el estudio diagnóstico y poder instaurar medidas preventivas precozmente. Asimismo, dado que la mayoría de los casos de MSC pueden tener un origen congénito y las principales etiologías presentan herencia autosómica dominante, los antecedentes familiares cobran gran relevancia en este grupo de pacientes (4). Un 25% de los casos de MSC se presentan durante el ejercicio, constituyendo así los deportistas de alto rendimiento un grupo de mayor riesgo (1,3).
La sobrevida de la MSC extrahospitalaria es de 20%. Esta cifra es más alta en países con mayor instrucción en técnicas de reanimación cardiovascular y disponibilidad de desfibriladores externos automáticos (DEA) en puntos estratégicos (1).
Epidemiología
A diferencia de la población adulta, la MSC es infrecuente en la edad pediátrica, con cifras aproximadas de 8,3 por 100.000 niños al año (2), siendo más común en hombres (2:1). Las etiologías más frecuentes se presentan en la Tabla 1 (5).

Enfrentamiento desde la Pediatría:
La American Heart Association (AHA) recomienda el uso de un cuestionario dirigido a la identificación de pacientes en riesgo, ya sea por elementos en la historia personal y antecedentes familiares, o por hallazgos de un examen físico acucioso con énfasis en lo cardiovascular (2) y que permite determinar que pacientes es preciso referir al subespecialista (Tabla 2). Esto permite discriminar con mayor precisión y seguridad entre pacientes, optimizar recursos y no sobresaturar la derivación al nivel terciario.
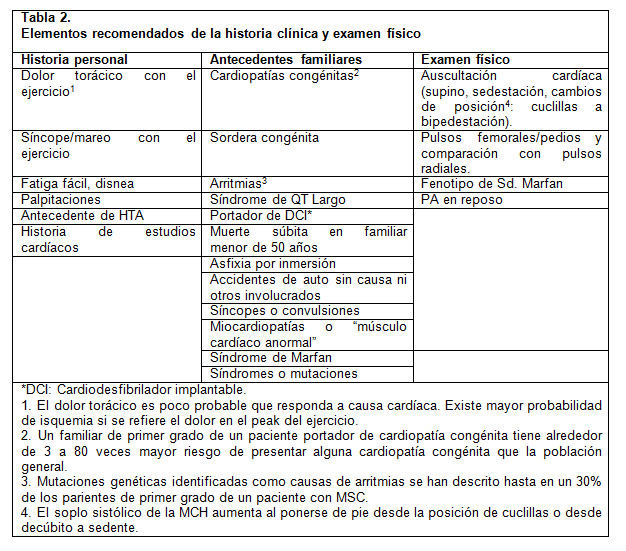
Con respecto al estudio con Electrocardiograma (ECG), la recomendación del equipo de Cardiología del Hospital Roberto del Río es utilizarlo como parte del screening en población pediátrica que practican deporte en forma competitiva.
Existen hallazgos electrocardiográficos que pueden ser indicadores de trastorno del ritmo que confiera al paciente mayor riesgo de presentar MSC, sin haber presentado síntomas previamente, como por ejemplo Sd. QT Largo, Sd. Brugada, Sd. Wolff Parkinson White. Sin perjuicio de lo anterior, existen condiciones que predisponen a la MSC que no se acompañan de alteraciones en el ECG, por lo que no es correcto asumir que un ECG normal excluye la posibilidad de presentar un evento cardiovascular y se debe recordar siempre la evaluación integral descrita previamente, asociando antecedentes personales, familiares, examen físico y la clínica de los pacientes.
Manifestaciones clínicas
Existen diversos síntomas y signos que se atribuyen habitualmente al sistema cardiovascular, si bien no necesariamente constituyen manifestaciones de alguna condición subyacente. Es fundamental la correcta interpretación de la clínica de los pacientes para orientar la causa, relacionar con el posible riesgo de MSC y también para tranquilizar e informar adecuadamente al paciente y su familia cuando la causa probable no es de origen cardíaco. Se profundiza a continuación con mayor detalle el concepto de síncope, dolor torácico y palpitaciones.
Síncope
Corresponde al compromiso de conciencia brusco, con pérdida del tono postural, autolimitado y con recuperación ad integrum (6). Es un motivo de consulta frecuente en la práctica clínica siendo alrededor del 3% de las consultas en SU pediátricas. En la literatura se describe que entre un 15 – 25% de la población presentará al menos un episodio antes de alcanzar la adultez (2), con mayor frecuencia de presentación en el sexo femenino y durante la adolescencia.
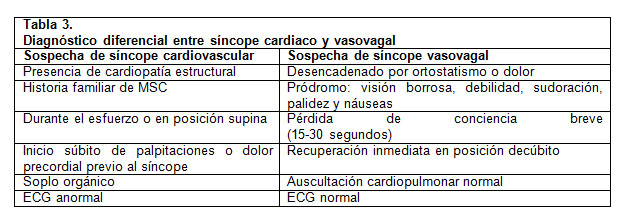
Alrededor de un 75% de los síncopes son de origen neurocardiogénico (vasovagal) y no revisten riesgo de muerte súbita cardíaca (6). Dentro de sus características principales se encuentran la presencia de síntomas prodrómicos y el cambio postural (desde decúbito o sedestación a bipedestación) previo al episodio. Se deben buscar aquellas características o “banderas rojas” que hagan sospechar un síncope cardiogénico, enumeradas en la Tabla 3 (5). Se debe destacar que aquellos síncopes que ocurren durante el ejercicio, en el agua o precedidos por dolor torácico y/o palpitaciones (2), deben motivar una evaluación urgente orientada a descartar patología cardiaca estructural o eléctrica.
Dolor torácico y palpitaciones
Ambos son motivo de consulta frecuente en pediatría. La gran mayoría de los dolores precordiales en la infancia responden a causas benignas o no cardíacas (95% de los casos) (7). Entre las principales etiologías se encuentran aquellas sin causa conocida (idiopáticas) las músculoesqueleticas; existiendo también causas respiratorias, gastrointestinales y psicógenas. El dolor precordial de causa cardiológica en la población pediátrica no supera el 5% de los casos y las principales etiologías son arritmias, anomalías estructurales y procesos inflamatorios o infecciosos (8).
Respecto a las palpitaciones, es relevante precisar características sobre el inicio, duración, contexto en que se presentan y la presencia de síntomas asociados para identificar aquellos episodios que con mayor probabilidad son de causa cardíaca, como lo son: inicio y término brusco, gatillado con el ejercicio, sonidos o emociones intensas, presencia de disnea, dolor precordial o síncope asociado (7). La mayoría de las veces el estudio con ECG o Holter de ritmo determina la presencia de arritmias o alteraciones que confieren el riesgo de presentarlas (2).
Se debe derivar a cardiólogo infantil a aquellos pacientes cuya historia presenta elementos que confieran mayor riesgo cardiovascular o de MSC (antecedentes familiares de MS o trastornos del ritmo, aparición del dolor con el peak de la actividad física, asociación de las palpitaciones con dolor torácico y/o disnea). Lo anterior, debido a que la posibilidad de cardiopatía estructural o trastorno del ritmo es mucho mayor en estos pacientes que en la población general.
Etiologías en la MSC
El evento final que determina la MSC tiende a ser una arritmia ventricular (taquicardia/fibrilación ventricular, torsión de puntas) o el colapso cardiovascular por bajo gasto cardíaco severo. Lo que varía entre los pacientes es la condición fisiopatológica de base que determina el evento.
En pediatría, los dos grandes grupos de enfermedades responsables de las causas primarias de la MSC son los defectos estructurales o funcionales y los defectos eléctricos (2). La MSC secundaria a consumo de drogas, la contusión torácica y la Hipertensión pulmonar primaria son etiologías raras en esta población. La Tabla 4 resume de las principales etiologías primarias y secundarias de la MSC (1). Posteriormente se presenta una breve reseña de las condiciones más frecuentes.
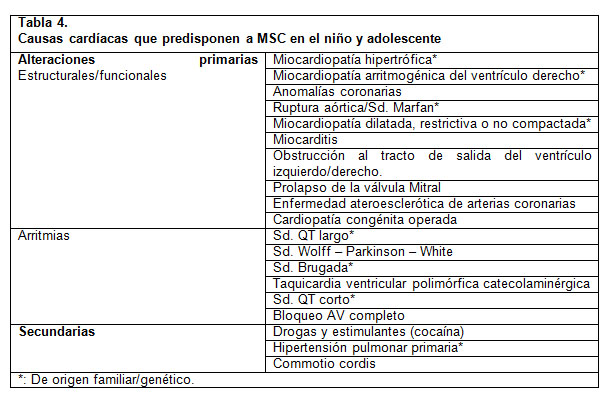
Defectos estructurales/funcionales:
Miocardiopatía hipertrófica (MCH):
Se define por la presencia de hipertrofia ventricular izquierda (HVI) en ausencia de condiciones que determinen cambios en los volúmenes de carga, obstrucciones al tracto de salida del ventrículo izquierdo (OTSVI) u otra alteración que determine la hipertrofia (9).
Es la principal causa de MSC en personas jóvenes. Tiene una prevalencia de hasta 1:500 en la población general y da cuenta de entre el 25 – 36% de los casos de MSC pediátrica (2), con mayor frecuencia en adolescentes de sexo masculino. Se debe a una mutación en genes de sarcómeros del miocardio que conducen a una remodelación, fibrosis e hipertrofia ventricular, con mayor compromiso del septum interventricular (9). La gran mayoría de los casos de MCH son de origen genético y se han descrito diversas mutaciones que se manifiestan con el fenotipo descrito, pero esta condición también se presenta en el contexto de enfermedades sistémicas, como por ejemplo la Enfermedad de Pompe (Glucogenosis tipo II).
La MSC en estos pacientes es secundaria a arritmias ventriculares (AV), con una mortalidad global de entre 2% y 6% por año. El sustrato arritmogénico estaría determinado por la fibrosis, la OTSVI y la isquemia por hipoperfusión relativa del músculo cardíaco hipertrófico (9).
La mayoría de los pacientes son asintomáticos, pudiendo ser la MSC la primera manifestación de la enfermedad. Entre los síntomas característicos, se presenta con disnea, angina o síncope con el ejercicio. El mecanismo fisiopatológico es la OTSVI por la hipertrofia septal, la cual empeora con el movimiento del velo septal de la válvula mitral hacia el TSVI (9).
Los hallazgos al examen físico varían en función de la severidad de la OTSVI. Es posible observar y palpar el choque de la punta prominente y lateralizado, pudiendo percibir un doble impulso en el choque o al palpar el pulso (“bisferiens”). La auscultación revela un soplo sistólico eyectivo en el foco aórtico, el que se amplifica con maniobras que disminuyan la precarga o aumenten la contractilidad (Valsalva, ejercicio) o al pasar de decúbito supino a posición sedente (al contrario de los soplos sistólicos inocentes) (9).
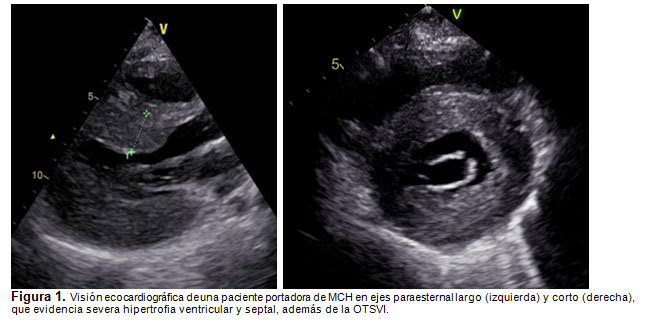
El estudio con ecocardiografía evidencia la hipertrofia septal y el grado de obstrucción al tracto de salida del VI (Fig. 1). El ECG puede presentar signos de hipertrofia ventricular izquierda, con S profundas en V1-V2 y R altas en V5-V6, desviación del eje a izquierda y ondas Q patológicas en V5-V6 y alteraciones del segmento ST (Fig. 2) (9). La RNM cardíaca permite definir el grado de compromiso ventricular (cicatrización) por su mejor resolución y complementa la estratificación del riesgo del paciente, pero no es necesaria para el diagnóstico (9).
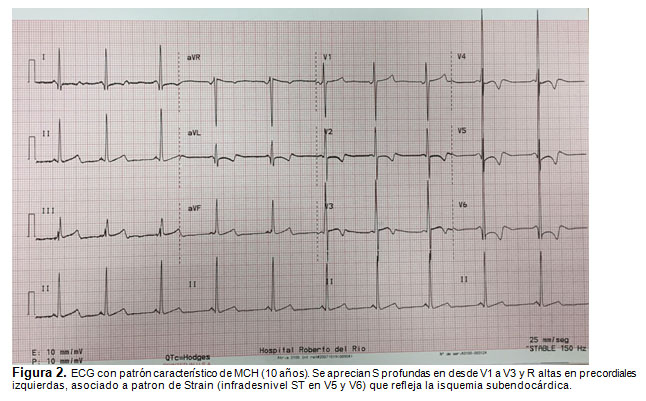
El manejo de la MCH está dado por la restricción del deporte competitivo, uso de betabloqueadores y bloqueadores de canales de Calcio (2,9). La indicación de uso de Cardiodesfibrilador implantable (DCI) se reserva para pacientes con riesgo aumentado de MSC: muerte súbita recuperada, taquicardia ventricular (TV) documentada, historia reciente de síncope, historia familiar de MSC asociada a MCH, hipertrofia septal mayor a 30 mm y una respuesta presora anormal durante el ejercicio (insuficiente incremento o franco descenso de la presión sistólica) (2).
El pronóstico varía en función de la severidad de la enfermedad y de la causa (genética, enfermedad metabólica, síndromes malformativos). En pacientes con mayor OTSVI, disfunción sistólica e insuficiencia cardíaca (IC) sintomática el pronóstico es peor, con mayor morbilidad y riesgo de MSC (9).
Es importante recalcar que el componente hereditario (autosómico dominante en la mayoría de los casos de MCH) conduce a que esta cardiopatía – que causa el mayor número de MSC en la población pediátrica y adulta joven – deba ser dirigidamente estudiada en todo familiar de primer grado de un paciente con MCH (y con mayor razón si existe el antecedente de MS o muerte súbita recuperada en la familia).
Anomalías coronarias
Constituyen la segunda causa (19%) de MSC en registros de autopsias de EEUU y pueden presentarse tanto en corazones estructuralmente normales o en el contexto de otras malformaciones cardíacas (2). Existen distintas variantes anormales en la emergencia de las arterias coronarias y no todas tienen significancia clínica, pudiendo ser formas asintomáticas o presentar MSC como primera manifestación (9). El origen anormal de una arteria coronaria puede conducir a isquemia miocárdica, sobre todo durante el ejercicio. La isquemia crónica y la cicatrización miocárdica resultante es el sustrato para la dilatación, disfunción y aparición de arritmias ventriculares potencialmente fatales (2). No presentan transmisión hereditaria.
Las anomalías pueden darse por un origen anómalo desde otro seno coronario, por un trayecto interarterial - entre la Aorta y la arteria Pulmonar (AP) - o un trayecto intramural (2). El origen anómalo de la coronaria izquierda desde el seno coronario derecho es el que asocia mayor riesgo de MSC, al punto de tener indicación quirúrgica incluso en pacientes asintomáticos con una coronaria de trayecto intramural (9,1).
La clínica varía según la alteración específica, pudiendo ser asintomáticos y el diagnóstico ser un hallazgo ecocardiográfico o manifestarse como MSC o falla cardíaca producto de dilatación y disfunción ventricular secundaria a la isquemia crónica de un miocardio dependiente de colaterales para su perfusión (9). En pacientes cuya anatomía (colaterales coronarias adecuadas) les permite desarrollarse sin evidencia de patología, la sintomatología clásica aparece durante el ejercicio y puede ser referida como angina, síncope, mareo o palpitaciones, debiendo tener alto grado de sospecha con los pacientes que refieran los síntomas en el peak del ejercicio y con mayor razón aquellos con MS recuperada durante el ejercicio.
El examen físico y la historia familiar pueden no aportar al diagnóstico, como también el ECG, que puede no mostrar alteraciones (2).
El estudio de elección es la ecocardiografía y puede ser complementado con Angiotac coronario o angiografía coronaria. Puede sospecharse mediante test de esfuerzo que presente alteraciones en el ECG (cambios en el segmento ST, ondas Q patológicas) (9). El manejo definitivo es la resolución quirúrgica.
Miocardiopatía Arritmogénica del Ventrículo Derecho (MAVD)
Enfermedad autosómica dominante, cuyo defecto genético genera una alteración estructural del ventrículo derecho, lo que conlleva a una dilatación del ventrículo, disfunción y un riesgo aumentado de arritmias ventriculares y de muerte súbita.
La clínica está definida por el grado de compromiso ventricular, la edad y el grado de actividad, destacando que es infrecuente que los pacientes presenten síntomas antes de los 13 años, a pesar de estar en riesgo de MSC (9). Alrededor del 10% de los pacientes portadores de MAVD presentarán MSC como primera manifestación de la enfermedad, pero la sintomatología más frecuente corresponde a palpitaciones y síncope con el ejercicio (2).
El examen físico puede ser normal y el diagnóstico se apoya en el ECG con ciertas alteraciones características. La ecocardiografía suele no presentar alteraciones en edades pediátricas y el gold standard en el diagnóstico es la resonancia magnética cardíaca (RMC), que permite caracterizar mejor el compromiso miocárdico.
El manejo consiste en la restricción del deporte competitivo y el uso de fármacos betabloqueadores y antiarrítmicos (2). Los pacientes portadores de MAVD pueden ser candidatos a DCI, una medida terapéutica efectiva para evitar la MSC y en casos de dilatación severa y disfunción, deben recibir el manejo farmacológico habitual de insuficiencia cardíaca (9).
Miocardiopatía dilatada (MCD) y Ventrículo Izquierdo No Compactado (VINC)
Son miocardiopatías menos frecuentes, con una prevalencia menor a 1:100.000. Presentan riesgo de MSC por arritmias ventriculares secundarias a la alteración estructural miocárdica (2).
En ambos casos la alteración estructural produce un deterioro severo de la función, por lo que se presentan con insuficiencia cardíaca, pero también con arritmias ventriculares y eventos tromboembolígenos. El diagnóstico en ambos casos se realiza con ecocardiografía.
Existen diversas medidas terapéuticas, considerando el uso de diuréticos de asa, betabloqueadores, inhibidores de la enzima convertidora de angiotensina (IECAs), antagonistas de Aldosterona y anticoagulantes.
En etapas avanzadas, las terapias de soporte mecánico (resincronizadores, dispositivos de asistencia ventricular) logran optimizar la clínica en pacientes con disincronía y el uso de DCI permite evitar eventos de MSC y prolongar la sobrevida. El trasplante cardíaco es una alternativa en casos graves de la enfermedad.
Miocarditis
Corresponde a una inflamación del miocardio, en la mayoría de los casos secundario a infección viral, pero también puede ser causada por bacterias, hongos o parásitos, además de reacciones de hipersensibilidad, autoinmunidad, toxinas y enfermedad de Kawasaki (9). Da cuenta del 7% de las MSC en atletas y es un hallazgo frecuente en necropsias de pacientes con muerte súbita de causa desconocida (1,2).
La miocarditis causa MSC por arritmias ventriculares, las que pueden ocurrir tanto en fase aguda como crónica de la enfermedad. El cuadro clínico característico - con el antecedente de un cuadro viral reciente – presenta taquicardia sinusal, aparición de un tercer ruido a la auscultación, troponinas elevadas y disfunción ventricular (2).
El diagnóstico se obtiene por la historia, examen físico, ecocardiografía y biomarcardores elevados. El ECG presenta taquicardia sinusal, voltajes disminuidos, aplanamiento o inversión de la onda T y alteraciones de la repolarización ventricular (9).
La mayoría de los pacientes (87%) evoluciona favorablemente. En casos de Miocarditis fulminante se puede requerir asistencia con ECMO o incluso trasplante cardíaco (2).
Cardiopatías congénitas
El riesgo de MSC en pacientes portadores de una cardiopatía congénita (CC) varía de entre 25 a 100 veces el de la población general y representa entre el 5 – 10% de las causas de MSC (1, 2, 11). Es más frecuente en CC cianóticas y lesiones obstructivas del corazón izquierdo. La MSC puede ser secundaria a arritmias, embolías o alteraciones circulatorias.
Cardiopatías congénitas que se asocian a mayor riesgo de MSC son: síndrome de corazón izquierdo hipoplásico, Tetralogía de Fallot, Transposición de grandes arterias (d-TGA), atresia pulmonar, estenosis aórtica y canal auriculoventricular. A su vez, los síndromes que asocian cardiopatías congénitas y que presentan mayor riesgo de MSC son: Sd. Down, Sd. Turner y Sd. Noonan (11).
Los pacientes portadores de Sd. Marfan, Sd. Ehlers Danlos y colagenopatías pueden presentar dilatación de la raíz de la Aorta y riesgo de disección en edades adultas, por lo que deben ser evaluados para definir el grado de dilatación y su manejo con betabloqueadores. El seguimiento ecográfico del compromiso aórtico es fundamental para definir el riesgo de disección y eventual indicación quirúrgica.
Arritmias
Síndrome de QT Largo (SQTL)
Trastorno asociado a múltiples mutaciones de canales de sodio y potasio, que se manifiesta en el ECG con un intervalo QT prolongado por una fase de repolarización retardada. Su prevalencia en la población es de 1: 2.500 (2) y su forma autosómica recesiva se relaciona con sordera neurosensorial congénita (Síndrome de Jervel y Lange-Nielsen). El SQTL tipo 1 es el más común de todos los fenotipos y junto al SQTL tipo 2 y tipo 3 representan aproximadamente al 75% de los casos de esta canalopatía (9).
La alteración de los canales iónicos genera una prolongación de la fase de repolarización, lo que aumenta el riesgo de FV o Torsión de puntas (TdP). Los gatillantes son el ejercicio y aumento del tono simpático (SQTL tipo 1), estímulo emocional y auditivo (SQTL tipo 2), bradicardia y sueño (SQTL tipo 3) (9).
La clínica varía desde el paciente asintomático hasta la MSC como primera manifestación. Si se gatilla una TdP, en función de la duración del episodio y la hipoperfusión cerebral, los pacientes pueden presentar episodios de palpitaciones, síncope o convulsiones (9).
El diagnóstico se basa en los criterios de Schwartz (2), que consideran antecedentes familiares, historia del paciente y el hallazgo en el ECG de un segmento QT corregido prolongado (> 450 ms) o la documentación de una arritmia de tipo Torsión de puntas. Cada tipo de SQTL presenta un patrón característico en el ECG, destacando en el SQTL tipo 1 ondas T de base amplia y picudas (Fig.3) (13). El test genético permite confirmar el diagnóstico y realizar el estudio familiar. Es fundamental el estudio de los familiares del paciente, por la posibilidad de pesquisar casos asintomáticos y realizar prevención primaria.
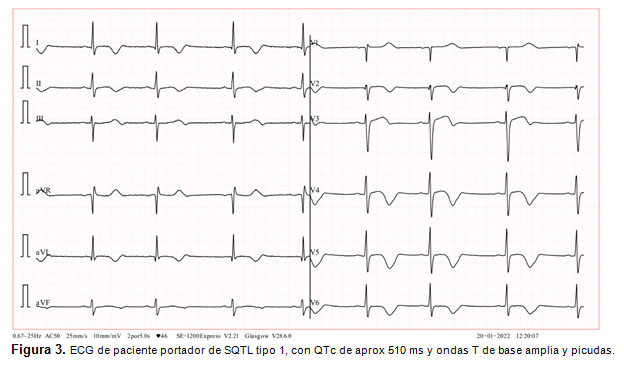
El manejo consiste en evitar los deportes competitivos, el uso de betabloqueo en pacientes con SQTL tipo 1 o 2 y la instalación de DCI en casos de muerte súbita recuperada y Torsión de puntas documentada (2, 9). Se debe recordar que existen diversos fármacos que prolongan el QT, que deberán ser evitados en estos pacientes. Se puede consultar sobre el efecto de diversos fármacos sobre el segmento QT (www.qtlongdrugs.org).
Manejo agudo y prevención de la muerte súbita cardíaca
El período de desfibrilación potencialmente efectiva corresponde a los primeros minutos desde ocurrido el paro cardíaco y la sobrevida se reduce un 10% por cada minuto en que se tarda en iniciar la reanimación cardiopulmonar (2). Éste es el principal motivo por el cual la sobrevida extrahospitalaria de la MSC no supera el 20% y se encuentra muy determinada por la educación en reanimación básica de la población y por la disponibilidad de desfibriladores externos automáticos (DEA) (1).
Dado que la causa cardíaca más frecuente de MS es la fibrilación ventricular, la posibilidad de recibir terapia eléctrica precoz es determinante en la sobrevida. En los casos en que el primer ritmo de paro cardíaco es potencialmente desfibrilable (TV o FV), el éxito de la reanimación avanzada es mayor que en aquellos casos no susceptibles de ser manejados eléctricamente (AESP y asistolia) (14).
La prevención de la MSC en pediatría es muy compleja y comprende múltiples variables. En el caso de la prevención primaria, hay que considerar que en un porcentaje de los casos la primera manifestación de enfermedad cardíaca puede ser el evento de muerte súbita (2). Lo anterior reafirma la necesidad de realizar un screening completo y dirigido, sobre todo en población de mayor riesgo (deportistas, parientes de primer grado de paciente fallecido por MSC o de un paciente portador de cardiopatía, canalopatía, etc) (2, 9). La variabilidad en el fenotipo de una misma condición genética (tanto por expresividad variable como por penetrancia incompleta de ciertas mutaciones), incrementan la dificultad para tomar una única conducta frente a los distintos diagnósticos, ya que no todos los pacientes serán sintomáticos o tendrán el mismo riesgo de MSC (4, 15).
El tratamiento farmacológico dirigido, cuando existe un diagnóstico específico, permite el manejo de síntomas y evitar progresión de la enfermedad o mejorar la función cardíaca con resultados variables, pero el tratamiento del evento mismo mediante un desfibrilador cardíaco implantable es lo que evita la muerte. En la población pediátrica el uso de DCI presenta más riesgos que en adultos, lo que dificulta la estandarización en su indicación en pediatría. Dificultades con el funcionamiento de cables, mayor frecuencia de descargas inapropiadas, complejidad de su implantación en pacientes pequeños son algunos de los factores que se deben considerar previo a la indicación de DCI en nuestra población de pacientes (9).
Existen condiciones en que se plantea el DCI, considerando la patología de base, la severidad de los síntomas del paciente, los hallazgos en el estudio electrocardiográfico y de imagen cardíaca que presente, los que se integran para generar un perfil de riesgo. En caso de MS recuperada, la indicación de DCI es categórica. Algunos diagnósticos en que se considera la DCI en población pediátrica para prevención primaria y secundaria son la MCH, SQTL, Sd. Brugada y CC (corregidas o no) asociadas a taquicardias ventriculares sostenidas sincopales que no son tratables farmacológicamente o con ablación (13, 16).
En todo caso de MSC o de diagnóstico de patologías con alto riesgo de MSC se debe realizar un screening en los familiares conocido como “cascada” (2, 4). Esto consiste en estudiar dirigidamente a los familiares directos y en el caso de identificar otro caso portador de la enfermedad, se debe reiniciar la búsqueda con sus familiares de primer grado, lo que se repite hasta que hayan sido estudiados todos los familiares potenciales portadores. Este proceso cobra mayor importancia en aquellos diagnósticos de conocida herencia autosómica dominante, siendo el mejor ejemplo las canalopatías en que se han descrito las mutaciones específicas de los canales iónicos afectados (SQTL, Sd. Brugada) (11).
Conclusiones
La muerte súbita cardíaca representa el 90% de los casos de MS en pediatría y alrededor de la mitad de los pacientes que fallecen por esta causa tiene historia de haber presentado síntomas cardiológicos o antecedentes familiares de riesgo para MSC. Es por esto que la identificación de los pacientes en riesgo debe ser una prioridad en la atención habitual en pediatría.
La sospecha y referencia al subespecialista permite realizar un diagnóstico precoz, lo que conduce a un tratamiento oportuno y seguimiento, con la posibilidad de realizar prevención primaria de MSC y además estudiar a los familiares para prevenir casos en portadores asintomáticos.
La recomendación es realizar en la consulta pediátrica habitual un screening dirigido a la pesquisa de elementos que aumenten el riesgo de padecer MSC, tanto de la historia personal, familiar y del examen físico cardiovascular. Por esto, es fundamental conocer que características clínicas sugieren patología cardíaca, que hallazgos al examen físico deben considerarse como patológicos y que antecedentes personales y familiares confieren al paciente mayor riesgo. Todo lo anterior permitirá una referencia oportuna al especialista.
En pediatría es frecuente la consulta por síntomas relacionados con patología cardiovascular (lipotimia, síncope, palpitaciones y dolor torácico), pero sólo un porcentaje menor de estos tiene su origen en una condición cardíaca. El reconocimiento de aquellos pacientes que realmente están en mayor riesgo de presentar MSC permite evitar la derivación excesiva o innecesaria al nivel terciario y además transmitir tranquilidad y educar a los padres sobre la salud de sus hijos.
Se debe recordar que cuando un paciente presenta síncope, dolor torácico y/o palpitaciones durante el ejercicio debe ser considerado como señal de alarma y requiere de una evaluación completa por cardiología. Además, todos los familiares de primer grado de pacientes fallecidos por muerte súbita cardíaca (o de causa desconocida) antes de los 50 años deben ser estudiados dirigidamente para descartar una condición subyacente que aumente el riesgo de MSC.
Referencias
- Picarzo L, Care P. Colaboración especial. 2015;77–86.
- Hammond BH, Zahka KG, Aziz PF. Sudden cardiac death: A pediatrician’s role. Pediatr Rev. 2019;40(9):456–65.
- Campbell R, Berger S, Ackerman MJ, Morrow WR, Jenkins K, Minich LLA, et al. Pediatric sudden cardiac arrest. Pediatrics. 2012;129(4).
- Gray B, Ackerman MJ, Semsarian C, Behr ER. Evaluation After Sudden Death in the Young: A Global Approach. Circ Arrhythm Electrophysiol. 2019 Aug;12(8): e007453. doi: 10.1161/CIRCEP.119.007453. Epub 2019 Aug 19. PMID: 31422686.
- Pérez Lescure J. Prevención de la muerte súbita cardíaca en pediatría; el insustituible papel del pediatra de Atención Primaria. 2015;159–66. Available from: www.aepap.org.
- Kanjwal K, Calkins H. Syncope in Children and Adolescents. Cardiol Clin [Internet].2015;33(3):397–409. Available from: http://dx.doi.org/10.1016/j.ccl.2015.04.008.
- Río R Del, Precordial D, Pediatría EN, Al P, Daniela D, Patricia D, et al. Revista Pediatría Electrónica Revista Pediatría Electrónica. 2018;15.
- Ji Hye Chun et al. Analysis of clinical characteristics and causes of chest pain in children and adolescents. Korean J Pediatr. 2015 Nov; 58(11):440-445.
- Wernovsky G et all. Anderson’s Pediatric Cardiology Robert H. Anderson, BSc, MD, PhD (Hon), FRCPath, FRCS Ed (Hon) Krishna Kumar, MD, DM. 2020;5114–42.
- Donoso FA, Cruces RP, Díaz RF, Clavería CC. Shock cardiogénico causado por implantación anómala de arteria coronaria. Rev Chil Pediatr. 2008;79(5):509–15.
- Mishra V, Zaidi S, Axiaq A, and Harky A (2020) Sudden cardiac death in children with congenital heart disease: a critical review of the literature. Cardiology in the Young 30: 1559–1565. doi: 10.1017/S1047951120003613.
- Cano-Hernández KS, Nava-Townsend S, Sánchez-Boiso A, Sánchez-Urbina R, Contreras-Ramos A, Erdmenger-Orellana JR, et al. Prevalence and spectrum of diseases that predispose to sudden cardiac death in mexican children: A sample obtained from the Federico Gomez children’s Hospital of Mexico. Arch Cardiol Mex. 2018;88(4):268–76.
- Albert Brotons DC. Cardiología pediátrica y cardiopatías congénitas del niño y del adolescente [Internet]. 2015. 555–60 p. Available from: http://www.secardioped.org/modules.php?name=webstructure&lang=ES&idwebstructure=21
- American Heart Association. Recursos para el manejo de emergencias circulatorias; Soporte Vital Av Pediatr. 2017;235–7.
- Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Vol. 138, Circulation. 2018. 272–391 p.
- Morell VO, Vetterly CG, Pedro J. Critical Care of Children with Heart Disease. Critical Care of Children with Heart Disease. 2020.
|
