La Hemoglobinuria Paroxística Nocturna también llamado Síndrome de Marchiafava-Micheli, es un desorden clonal manifestado por anemia hemolítica intravascular por activación del complemento1, 2, provocando trombosis, daño del órgano terminal, falla medular.3
Se considera una enfermedad huérfana cuya incidencia se estima en 1/200.000 y prevalencia de 15.9 por millón, tiene un pico de presentación entre la 3ra y 4ta década de la vida, el 10 % se diagnostican antes de los 21 años, no hay predisposición racial, género ni familiar 4. En México se ha reportado una incidencia de 2 casos por 100,000 habitantes con presentación entre los 17 y 77 años siendo alrededor del 18% en edades pediátricas.5
La hemólisis en HPN está mediada por complemento y es resultado de una mutación somática del 1-6 N acetil glucosaminil transferasa del gen PIG-A (Phosphatydilinosotol Glycan Class A) de la célula madre hematopoyética localizado en el cromosoma Xp22.1, como consecuencia hay un déficit de GPI produciendo una alteración de las proteínas de anclaje de membrana principalmente el CD59 (MIRL) inhibidor de la lisis reactiva de membrana y CD55 (DAF) factor acelerador de la degradación incrementando su sensibilidad a las células hematológicas al complemento6,7,8. Se han descrito mutaciones adicionales como son HMGA2, mutación concomitante JAK2V617F, N-RAS mutante y una mutación concomitante HPN y BCR-ABL sin embargo estos casos parecen ser la excepción y no la regla.9
Existen dos teorías sobre la causa de HPN
- Teoría de la patogénesis dual: resulta precisamente de la coexistencia de 2 factores: el fallo de la médula ósea normal, con una mutación somática del gen PIG-A. El fallo de la médula ósea favorece el desarrollo del clon HPN el cual se expande como resultado de una selección negativa contra el stem cell hematopoyético normal.7 En consecuencia, la mayoría de la hematopoyesis consistirá en células deficientes en proteínas ligadas al GPI.10
- Teoría de escape: la expansión del clon HPN depende de la existencia de uno o más factores ambientales externos, los cuales ejercen una presión selectiva a favor del clon HPN. Uno de estos factores podría ser una injuria a las células hematopoyéticas normales, lo cual salva a las células HPN anormales.11 Existe evidencia para apoyar ésta hipótesis pero no puede explicar las siguientes observaciones: 1) PIG-A se expresa de forma ubicua en el cuerpo ¿por qué debería restringirse el ataque inmunitario contra el ancla?; 2) la terapia inmunosupresora no conduce a la eliminación de las células mutantes ni a la expansión del HSC normal con el retorno a la hematopoyesis normal ; y 3) una fracción significativa de pacientes con HPN sufre la extinción “espontánea” del clon.12
La expresión clínica depende del tipo de proteína de membrana (Tabla 1). Se caracteriza por la triada: 1) Hemólisis intravascular; 2) Trombosis y 3) Citogenias y falla en médula ósea.

- Hemólisis intravascular se produce por mayor sensibilidad al complemento con activación de complejo de ataque de membrana principalmente en las proteínas de membranas en los eritrocitos (CD55 y CD59) produciendo hemoglobina libre. Dependiendo del tipo de mutación en el gen PIGA, pueden ocurrir varios grados de deficiencia y esto contribuye a la variabilidad de la hemólisis en los pacientes. Clínicamente se observa dolor abdominal, disnea, disfagia, distonías del músculo liso, fatiga y hemoglobinuria, las exacerbaciones de la hemólisis severa puede resultar en insuficiencia renal.13
- La trombosis principal causa de mortalidad (40-60%), se han postulado búsquedas relacionadas con varias teorías para explicar la hipercoagulabilidad, pero el mecanismo aún no es claro. Se cree que la trombofilia en HPN está relacionada con el grado de hemólisis y, por lo tanto, indirectamente con el tamaño del clon de HPN. Las posibles vías protrombóticas incluyen la activación plaquetaria por componente del complemento, micropartículas procoagulantes derivadas de eritrocitos deficientes en GPI, o la desaceleración de la microcirculación debido a la vasoconstricción inducida por productos de hemólisis. También se ha sugerido que la hemólisis intravascular expone los fosfolípidos de los glóbulos rojos que pueden servir para iniciar la coagulación. Puede desarrollarse en territorio arterial o venoso, los sitios más comunes son intra-abdominal (hepático, portal, esplénico o mesentérica) y cerebral (seno sagital y cavernoso); la trombosis de la vena hepática (conocido como síndrome de Budd-Chiari) es el más común. Trombosis venosa profunda, embolia pulmonar y trombosis dermal.14
- Alteraciones hematólogicas depende del grado de afección en la médula ósea. Pancitopenia (32%), anemia (33%) y bicitopenia (17% - anemia y trombocitopenias) 13 Otros síntomas son disfagia, impotencia, sensación de fatiga, insuficiencia renal aguda y crónica, mayor incidencia a infecciones. Los componentes de la médula ósea de HPN puede presentarse desde una forma subclínica hasta anemia aplásica severa15
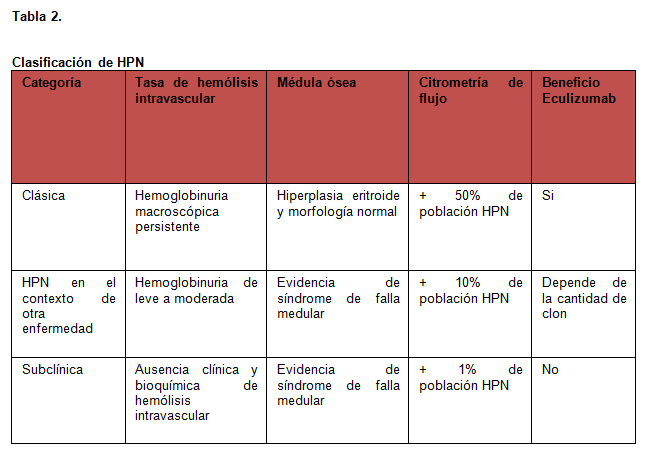
La clasificación de HPN se propuso por el Grupo Internacional con interés en HPN en: clásica, HPN en el contexto de otra enfermedad y subclínica (Tabla 2)
El diagnóstico diferencial se debe realizar en todo paciente quien presenta: Hemoglobinuria; Anemia hemolítica Coombs negativa; Trombosis venosa de localización inusual (síndrome de Budd Chiari, mesentérica, eje portal, cerebrales), con hemólisis; Disfagia intermitente o dolor abdominal de etiología no aclarada con evidencia de hemólisis; Aplasia medular (al diagnóstico y anualmente); Síndrome mielodisplásico hipoplásico; Citopenias idiopáticas y mantenidas de significado incierto.16
El diagnóstico de laboratorio se basa en la demostración de la sensibilidad de los eritrocitos al complemento, esto se realiza mediante las siguientes pruebas:
- Prueba de Ham: constituye la susceptibilidad de los eritrocitos HPN a la lisis en suero humano acidificado. Su eficacia es limitada, porque puede dar resultados falsos negativos por el efecto de transfusiones previas y falsos positivos en la anemia diseritropoyética congénita tipo II o HEMPAS. Actualmente no se realiza.17
- Prueba de la Sucrosa: la base de esta prueba es la fijación del complemento mediante la disminución de la potencia iónica del medio ambiente, que provoca la hemólisis exagerada de los eritrocitos HPN. Este examen es sensible, pero poco específico.
- Citometría de flujo; demuestra la alteración de las proteínas de anclaje en los granulocitos (CD16+/- CD66b +/- CD24) y al menos una línea celular adicional ya sea Glóbulos rojos (CD59) o Monocitos (CD14), se da el diagnóstico cuando se demuestra el defecto en al menos 2 líneas hematopoyéticas distintas de al menos dos marcadores (dos proteínas asociadas a GPI o una proteína asociada a GPIy FLAER) distintas. Células HPN encontradas son I: GPI-APs normales; II: Parcialmente deficientes de GPI-APs (90%) y III: Completamente deficientes de GPI-APs. Los fenotipos encontrados I y III: Más común (78%); I, II y III: Segundo más común y I y II: Menos frecuente.18,19
La variabilidad de las manifestaciones clínicas hace necesario individualizar el tratamiento, teniendo tres puntos importantes: 1) Corrección de la anemia; 2) Prevención y tratamiento de la trombosis; y 3) Modificación de la hematopoyesis. Actualmente se ha utilizado el Eculizumab es un anticuerpo monoclonal con alta afinidad a C5a y la formación C5b-9, está indicado en el tratamiento de pacientes con hemoglobinuria paroxística nocturna (HPN) para reducir la hemólisis y el tratamiento de pacientes con síndrome urémico hemolítico atípico (SHU) para inhibir la microangiopatía trombótica mediada por el complemento.20
El TCPH es la terapia curativa y está indicado en Aplasia medular grave + HPN, complicaciones graves (Tromboembolismos graves y/o recurrentes y Anemia transfusión dependiente), pacientes sin respuesta a Eculizumab y aquellos con evolución clonal (SMD, LMA), actualmente se sugiere un acondicionamiento no mieloabaltico de intensidad reducida con CSA + GALT + CFM reportando una sobrevida del 50-60%.21
Tienen una sobrevida sin tratamiento de 10 a 15 años. El 25% sobreviven 25 años o más al momento del diagnóstico. Hay factores pronósticos adversos: presencia de trombosis, edad mayor de 55 años y el desarrollo de otras mielopatías clonales, (síndrome mielodisplástico y la leucemia aguda). Los pacientes con diagnóstico de HPN clásico con leucopenia, trombocitopenia u otras complicaciones durante el tratamiento pueden disminuir su sobrevida. Dentro de su evolución la mayoría tiene un curso crónico, el 20% el HPN se diagnóstica posterior a AA, el 30% HPN precede a la AA, el 5% desarrolla SMD, el 1% tiene progresión leucémica, el 40% desarrollan trombosis siendo la principal causa de muerto el síndrome de Budd-Chiari.22
Conclusión
HPN es una enfermedad sistémica grave, rara y clínica heterogénea, la hemoglobinuria poco frecuente. Existen diferentes enfoques terapéuticos que no curan la enfermedad pero pueden disminuir sus complicaciones sin embargo el TCPH se considera el único tratamiento curativo en paciente con AA.
Bibliografía
- R. A. Brodsky, “Paroxysmal nocturnal hemoglobinuria,” 2014. Blood;124 (18): 2804–2811
- Dharshana Krishnaprasadh, Inna Kaminecki, Anna Sechser Perl, and Jonathan TeitelbaumParoxysmal Nocturnal Hemoglobinuria: Diagnostic Challenges in Pediatric Patient. Case Rep Pediatr. 2019; 2019: 4930494
- Brodsky R. A. Paroxysmal nocturnal hemoglobinuria. Blood. 2014; 124(18):2804–2811.
- Nathaniel Mon Père, Tom Lenaerts, Jorge M. Pacheco, David Dingli. Evolutionary dynamics of paroxysmal nocturnal hemoglobinuria. PLoS Comput Biol. 2018 Jun; 14(6): e1006133.
- Jesús Hernández-Reyes, Mónica Patricia González-Ramírez et al. Paroxysmal nocturnal hemoglobinuria in México: a 30-year, single institution experience. Rev Invest Clin. 2014; 66 (1): 12-6
- Parker CJ. Historical aspects of paroxysmal nocturnal haemoglobinuria: defining the disease. Br J Haematol 2002;117:3-22
- Salvatrice Mancuso, Giuseppe Sucato, Melania Carlisi, et al. Paroxysmal nocturnal hemoglobinuria: When delay in diagnosis and long therapy occurs. Hematol Rep. 2018 Mar 2; 10(1): 7523
- Stanley Chun-Wei Lee and Omar Abdel-Wahab.The mutational landscape of paroxysmal nocturnal hemoglobinuria revealed: new insights into clonal dominance. J Clin Invest. 2014 Oct 1; 124(10): 4227–4230
- Lucio Luzzatto. Recent advances in the pathogenesis and treatment of paroxysmal nocturnal hemoglobinuria. Version 1. F1000Res. 2016
- Gargiulo L, Lastraioli S, Cerruti G, Serra M, Loiacono F, Zupo S, et al. Highly homologous T-cell receptor beta sequences support a common target for autoreactive T cells in most patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007; 109(11):5036–42
- Amrallah A. Mohammed, Hani EL-Tanni, et al. Paroxysmal Nocturnal Hemoglobinuria: From Bench to Bed. Indian J Hematol Blood Transfus. 2016 Dec; 32(4): 383–391
- Shen W, Clemente MJ, Hosono N, Yoshida K, Przychodzen B, Yoshizato T, et al. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest. 2014; 124 (10):4529–38
- Milanes Roldan, María Teresa et al. Hemoglobinuria paroxística nocturna: Actualización. Rev Cubana Hematol Inmunol Hemoter. 2003;19 (1):2019-2022
- Malato A, Saccullo G, Lo Coco L, et al. Thrombotic complications in paroxysmal nocturnal haemoglobinuria: a literature review. Blood Transfus 2012;10: 428-35
- Johnson RJ, Hillmen P. Paroxysmal nocturnal haemoglobinuria: Nature’s gene therapy? Mol Pathol. 2002 Jun;55(3):145-52
- Prabhu Manivannan, Ankur Ahuja, and Hara Prasad Pat. Diagnosis of Paroxysmal Nocturnal Hemoglobinuria: Recent Advances. Indian J Hematol Blood Transfus. 2017 Dec; 33(4): 453–462
- Charles J. Parker. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2016 Dec 2; 2016(1): 208–216.
- Rodolfo Patussi Correia, Laiz Cameirão Bento, et al. Technical advances in flow cytometry-based diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria. Einstein (Sao Paulo). 2016 Jul-Sep; 14(3): 366–373.
- Sang Hyuk Park, M.D., Ph.D., Joseph Jeong. Comparison of High Sensitivity and Conventional Flow Cytometry for Diagnosing Overt Paroxysmal Nocturnal Hemoglobinuria and Detecting Minor Paroxysmal Nocturnal Hemoglobinuria Clones. Ann Lab Med. 2019 Mar; 39(2): 150–157
- Konar M, Granoff DM. Eculizumab treatment and impaired opsonophagocytic killing of meningococci by whole blood from immunized adults. Blood. 2017 Aug 17; 130(7):891–9
- Antonio M. Risitano, Serena Marotta, Patrizia Ricci, et al. Anti-complement Treatment for Paroxysmal Nocturnal Hemoglobinuria: Time for Proximal Complement Inhibition? A Position Paper From the SAAWP of the EBMT. Front Immunol. 2019; 10: 1157
- Goker H, Uz B, Buyukasik Y, Aksu S, Haznedaroglu I, Sayınalp N, Karacan Y, Tekin F, Ozcebe OI. Eculizumab before and after allogeneic hematopoietic stem cell transplantation in a patient with paroxysmal nocturnal hemoglobinuria: Case report. Turk J Haematol. 2011;28:223–7
|
