Introducción
El síndrome de Alagille (SALG) displasia arteriohepática, es una condición multisistémica autosómica dominante altamente variable con una frecuencia estimada de 1 en 30,000-100,000 dependiendo la población. Inicialmente se describió como una enfermedad hepática, pero las pruebas moleculares han demostrado que la afectación hepática es variable, incluida la ausencia de enfermedad hepática. Los casos que presentan enfermedad hepática significativa se presentan en el período neonatal con hiperbilirrubinemia conjugada. La tasa de mortalidad es del 10% por accidentes vasculares, enfermedad cardíaca y enfermedad hepática.1
Aproximadamente el 95% de los pacientes con SALG tienen mutaciones en JAG1, que codifica el ligando de la vía de señalización NOTCH Jagged-1. Las interacciones de Jagged/Notch que ocurren en los puntos de contacto célula-célula determinan el destino de la célula en el desarrollo temprano. Se encontró que las mutaciones NOTCH2 causaban SALG en pacientes que no tenían mutaciones en JAG1 y la enfermedad renal es más común en pacientes con defectos NOTCH2 que JAG1. 1
El SALG principalmente es un diagnóstico clínico que consiste en la escasez de conductos biliares intralobulares y al menos 3 de 5 características clínicas importante como: colestasis, enfermedad cardíaca con estenosis pulmonar periférica, anomalías esqueléticas con vértebras torácicas en alas de mariposas, embriotoxón posterior y facies triangular.1,2
Resultado
Paciente de 2 años procedente de zona rural, fruto de tercera gestación de madre de 23 años con antecedentes de labio leporino y paladar hendido, parto vaginal domiciliario, sin atención médica. Pobres controles prenatales, con exámenes para TORCH y otras infecciones negativas. Fue llevado a urgencias por episodio de hematemesis, dificultad respiratoria, distención abdominal y melenas escasas de un día de evolución. Paciente con antecedentes de colestasis desde el mes de nacido, baja talla y peso, sin hitos adecuados del neurodesarrollo para la edad. Dentro de la revisión de historia clínica refiere primo materno con hipotonía congénita con facies dismórficas y retraso del desarrollo psicomotor, sin diagnóstico establecido.
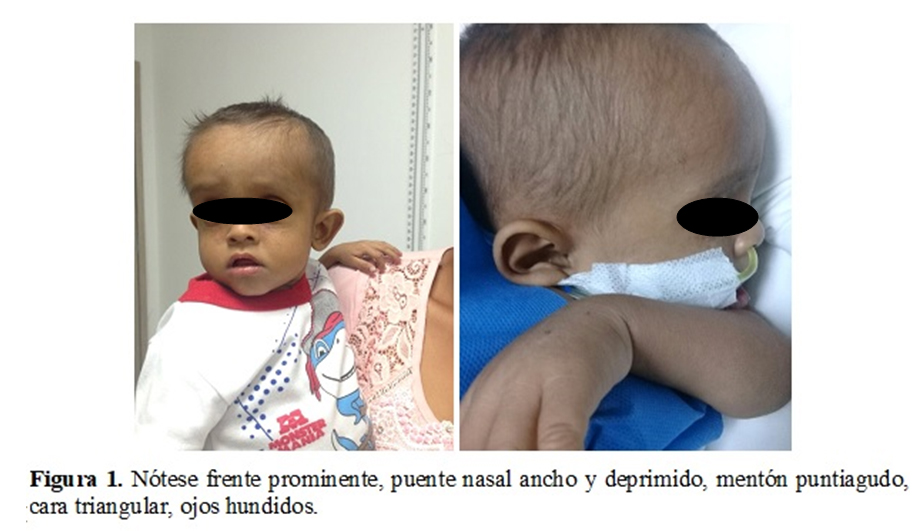
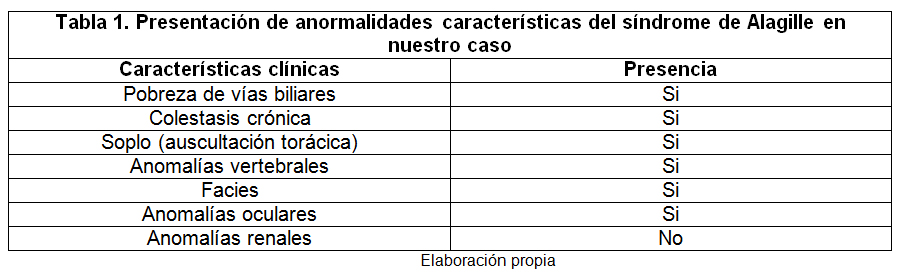
Al examen físico bajo peso y talla para la edad (Percentil 3). Llama la atención facies con facies dismórfica. (Figura 1. y Tabla 1.). Soplo sistólico grado II/VI de predominio pulmonar con hallazgo en ecocardiograma de estenosis leve de las arterias pulmonares. Hepatomegalia de 8 cm por debajo del reborde costal y ascitis. Se realizan exámenes de ingreso encontrando datos positivos como anemia severa, leucocitosis, transaminitis, hipoalbuminemia e hiperbilirrubinemia a expensas de bilirrubina directa y dislipidemia.
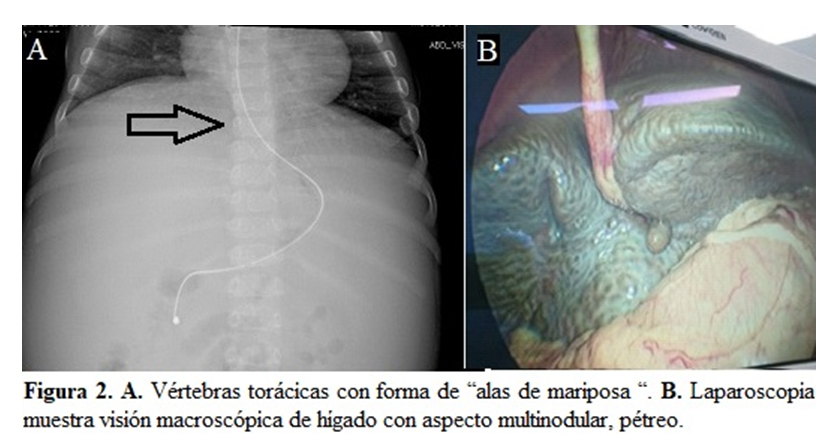
Se realizan estudios complementarios, ultrasonografía abdominal con hepatomegalia y esplenomegalia con patrón micronodular y radiografía de tórax en donde se evidencia deformidad en alas de mariposa de las vértebras torácicas sin otros hallazgos positivos en la serie de huesos largos (Figura 2.). Se realiza doppler de vasos portales normales y endoscopia de vías digestivas con hallazgo de dilataciones varicosas serpentiformes a 15 cm de la arcada dental y várices esofágicas menores de 5 mm que ocupaban el 30 % de la luz.
Se inicia protocolo de estudio para colestasis y manejo con colestiramina, propanolol, vitaminas y atorvastatina. Se solicita perfil TORCH, Epstein – Barr, gamma glutamil transpeptidasa y perfil metabólico. Se solicita biopsia hepática, encontrando a nivel macroscópico hígado multinodular pétreo color verdoso, con vesícula biliar de paredes delgadas. (Figura 2.) En la patología se observa disminución de los ductos biliares con proliferación ductular compensatoria. Por otro lado, el servicio de oftalmología al examen encuentra depósitos lipídicos perilimbares corneales superiores en ambos ojos, palidez extrema de papilas bilateral con atenuación vascular y coroidosis severa.
Luego del inicio del tratamiento médico, el niño evoluciona hacia la mejoría clínica. Valorado por el servicio de Genética solicitando panel para colestasis, con resultados positivos para variante patogénica detectada en el GEN JAG1.
Comentarios
El síndrome de Alagille es una enfermedad colestásica crónica de origen genético de carácter autosómico dominante. Se estima una frecuencia de 1 :30.000 niños nacidos.1,2 El trastorno de tipo genético involucra a dos genes JAG1 (98% de los casos ) y NOTCH2 ( 1 – 2 %), ubicados en el cromosoma 20 y 1 respectivamente, la explicación es una codificación de proteínas anormales que causan en general una disminución en la señalización NOTCH. La consecuencia principal de estas alteraciones es una ductupenia y ramificación defectuosa del árbol biliar que ocurre en el primer trimestre de la gestación hacia las 12 semanas, en donde los hepatoblastos periportales forman la placa ductal. 3,4
La colestasis neonatal ocurre en el 95% de los casos, pero puede ser leve y sin manifestaciones clínicas aparente. Cuando la colestasis es severa, puede ser clínicamente indistinguible de la atresia de vías biliares. Las características clínicas de los pacientes con este síndrome pueden ser muy variables, observando afectación cardiaca, hepática, oftalmológica, esquelética, renal y de facies peculiares. 5,6,7
En el caso reportado, la colestasis y hallazgos faciales (Figura 1. y Tabla 1.), provocaron la sospecha clínica realizando estudios que confirmen el diagnóstico. En cuanto a las características cardiacas más del 90% de los casos tienen anomalías, siendo la estenosis de las ramas pulmonares la más frecuente. Otras lesiones cardiacas encontradas en el 16% de los casos es la tetralogía de Fallot y los defectos septales en menor proporción.8,7
Los hallazgos en el sistema esquelético las vértebras torácicas en forma de alas de mariposa se deben a fusiones anormales de la columna vertebral que conducen a hendiduras sagitales en el 80% de los casos, tal como se evidencia en el caso expuestos. Otros hallazgos menos frecuentes son la craneosinostosis, sinostosis radioulnar y acortamiento de falanges distales de los dedos.
El 28% de los paciente pueden sufrir de fracturas en huesos de extremidades inferiores.8,9 EL hallazgo oftalmológico característico es el embriotoxón posterior en un 90% de los casos, pero hasta el 15% de los casos puede ser normal en la población de pacientes con defectos en el 22 q11. El crecimiento y la nutrición son vitales para el pronóstico de estos niños; la deficiencia de vitaminas liposolubles y el retraso de la pubertad son comunes, por lo cual los requerimientos de suplementos alimenticios, vitamina D, E, K,A son obligatorios.9
Los hallazgos oftalmológicos de base el más frecuente el embriotoxón posterior (79-89%) son muy comunes, sin llegar a afectar la agudeza visual. Otros defectos oculares que pueden llegar a presentarse incluyen la anomalía de Axenfeld-Rieger y policoria. La morbilidad de los pacientes con Alagille, se basa en otros hallazgos de tipo vascular, presentándose hasta en 15% accidentes neurovasculares y aneurismas intracraneales. El diagnóstico de esta enfermedad se basa en hallazgos clínicos, paraclínicos con presencia de hiperbilirrubinemia directa y alteraciones de la función hepática.10 Confirmación de anomalías cardiacas, oftalmológicas, renales y óseas que nos permitan descartar otras alteraciones. La confirmación de defectos genéticos es indispensable para la confirmación del diagnóstico, realizando la prueba para JAG1 y NOTCH2 prueba realizada en el caso en mención.10,11
El tratamiento de base para los pacientes con colestasis crónica se basa en la suplementación dietética adecuada, con una ingesta de proteínas de hasta de 4 gramos/kilos, aumento de la necesidad de aminoácidos de cadena ramificada tipo leucina e isoleucina y valina, complejos de la vitamina B, C y Zinc además de las liposolubles. Los bajos niveles de estas vitaminas pueden producir serios problemas en los ojos, nervios, músculos, huesos y factores de la coagulación. Se deben monitorear dichos niveles sanguíneos de estas vitaminas, ajustando las dosis cuando sea necesario.11,12
La colestasis produce acumulación en el cuerpo de bilirrubina, sales biliares y colesterol. Los medicamentos tales como el ácido ursodesoxicólico (ursodiol) pueden ayudar a mejorar el flujo biliar y disminuir el prurito. Ursodiol también ayuda a reducir los niveles de colesterol e ictericia en la sangre. La acumulación de las sales biliares puede causar prurito, algunas veces severo. Los antihistamínicos (tales como difenhidramina e hidroxicina) pueden ser usados para controlar el prurito y mejorar el sueño. Muchos pacientes son tratados con colestiramina y colesevelam para ayudar a remover las sales biliares del cuerpo. En casos muy severos, para ayudar con el prurito se utiliza la cirugía para remover el exceso de bilis (desvío biliar externo y parcial o exclusión ileal). 11,13
No hay una cura conocida para SALG, pero hay formas de prevenir o reducir los problemas de salud relacionados con SALG. El tratamiento para los niños con SALG es individualizado, pero la mayoría de los pacientes requieren tratamiento para la enfermedad hepática. La mortalidad global es de aproximadamente 10-30% de los casos se deben a falla cardiaca o falla hepática severa. El pronóstico a largo plazo depende de la severidad del daño hepático y las malformaciones asociadas. 12 Entre el 21-31% de los pacientes requiere trasplante hepático por el desarrollo de cirrosis como en nuestro caso. Otro 15% puede desarrollar hepatocarcinoma e insuficiencia hepática, pancreática, fibrosis hepática e hipertensión portal.13,14,15
El caso presentado demuestra que el diagnóstico tardío ocasiona daños irreversibles en la función de los órganos, por lo cual el tratamiento se basa en medidas de soporte y seguimiento interdisciplinario.
El Síndrome de Alagille es un trastorno poco frecuente con progresión variable de los síntomas y disfunción hepática. Los pacientes con insuficiencia hepática como resultado de la colestasis requieren un trasplante hepático para el tratamiento. La sospecha clínica en el diagnóstico de SALG es de gran importancia dándose hallazgos característicos con los criterios clásicos. Sin embargo, el diagnóstico puede ser muy difícil en pacientes en los que no cumplan con los criterios clásicos lo cual no fue nuestro caso. Por lo tanto, es una afección que presenta desafíos para el juicio clínico y se deben tener en cuenta las principales pruebas diagnósticas. El tratamiento es individualizado y debe ser interdisciplinario, las intervenciones quirúrgicas como trasplante de hígado es fundamental para la disminución de morbimortalidad.
Bibliografía
- Jane L. Hartley, Paul Gissen, Deirdre A. Kelly, Alagille Syndrome and Other Hereditary Causes of Cholestasis, Clin Liver Dis. 2013; 17: 279–300
- Turnpenny, P. D. & Ellard, S. Alagille syndrome : pathogenesis , diagnosis and management. 2011; 20: 251–257
- Dědič T, Jirsa M, Keil R, Rygl M, Šnajdauf J, Kotalová R. Alagille Syndrome Mimicking Biliary Atresia in Early Infancy. Alpini G, ed. PLoS ONE. 2015;10(11):e0143939.
- Ortega Pérez SN, González Santana D, Ramos Varela JC, Cañizo Fernández D, Peña Quintana L. Síndrome de Alagille; una patología que tener en cuenta. Rev Pediatr Aten Primaria. 2017;19:267-70.
- Sheflin-Findling S1, Arnon R, Lee S, Chu J, Henderling F, Kerkar N, Iyer K. Partial internal biliary diversion for Alagille syndrome: case report and review of the literature. J Pediatr Surg. 2012 Jul;47(7):1453-6.
- D'amato G, Mónica; Ruiz N, Patricia; Aguirre R, Karen And Gomez Rojas, Susana. Colestasis en pediatría. Rev Col Gastroenterol. 2016; 31(4): 409-417.
- Lee HP, Kang B, Choi SY, Lee S, Lee S-K, Choe YH. Outcome of Alagille Syndrome Patients Who Had Previously Received Kasai Operation during Infancy: A Single Center Study. Pediatric Gastroenterology, Hepatology & Nutrition. 2015;18(3):175-179.
- Saleh, Maha, Binita M Kamath, and David Chitayat. “Alagille Syndrome: Clinical Perspectives.” The Application of Clinical Genetics 9 (2016); 9: 75–82.
- Tirado-Pérez IS, Sequeda-Monterroza JF, Zarate-Vergara AC. Craneosinostosis: Revisión de literatura. Rev Univ. salud. 2016;18(1):182-189.
- Mirta Cioccaa, Fernando Álvarez, Síndrome de Alagille, Arch Argent Pediatr 2012;110(6):509-515
- Spinner NB, Leonard LD, Krantz ID. Alagille Syndrome. 2000 May 19 [Updated 2013 Feb 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1273/
- Khalaf, R., Phen, C., Karjoo, S. & Wilsey, M. Cholestasis beyond the Neonatal and Infancy Periods. 2016; 19: 1–11
- Kim J, Yang B, Paik N, Choe YH, Paik Y-H. A case of Alagille syndrome presenting with chronic cholestasis in an adult. Clinical and Molecular Hepatology. 2017;23(3):260-264.
- Jagadisan, B. & Srivastava, A. Child with Jaundice and Pruritus : How to Evaluate ? Indian J Pediatr (2016). 83(11), 1311–1320
- Kronsten V1, Fitzpatrick E, Baker A. Management of Cholestatic Pruritus in Paediatric Patients with Alagille Syndrome; A Review of 15 Years Experience at King's College Hospital. J Pediatr Gastroenterol Nutr. 2013 Aug;57(2):149-54.
|
