Síndrome GAPO, a propósito de un caso
José María García-Sánchez1, Laura Ibáñez Beltrán2, Eva López Blanco3, Patricia Gutiérrez Ontalvilla3
1 Médico Interno Residente Servicio de Cirugía Plástica, Hospital Universitari i politècnic la Fe, Valencia (España)
2 Médico Interno Residente Servicio de Pediatría, Hospital Universitari i politècnic la Fe, Valencia (España)
3 Médico Adjunto Servicio de Cirugía Plástica, Hospital Universitari i politècnic la Fe, Valencia (España)
Resumen | Abstract | Texto completo| Descargar cuerpo en pdf |
Introducción
El síndrome GAPO es un raro trastorno genético con fenotipo característico, cuyos rasgos clínicos afectan a un reducido número de personas a nivel mundial(1). Su singularidad y repercusión clínica, con afectación de múltiples aparatos, convierten a esta patología en un auténtico reto terapéutico.
Caso clínico
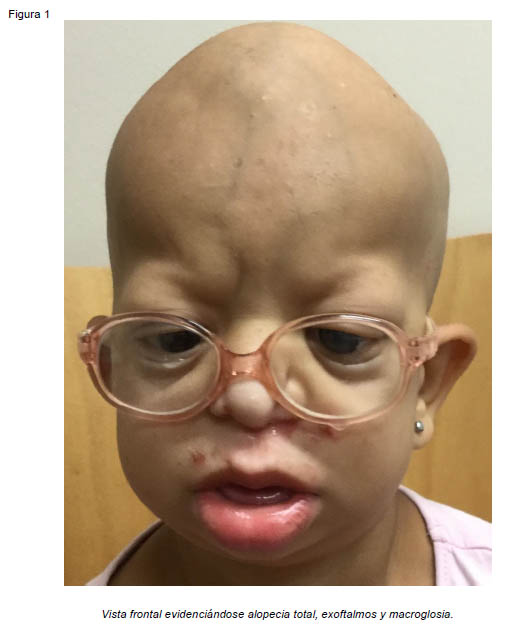
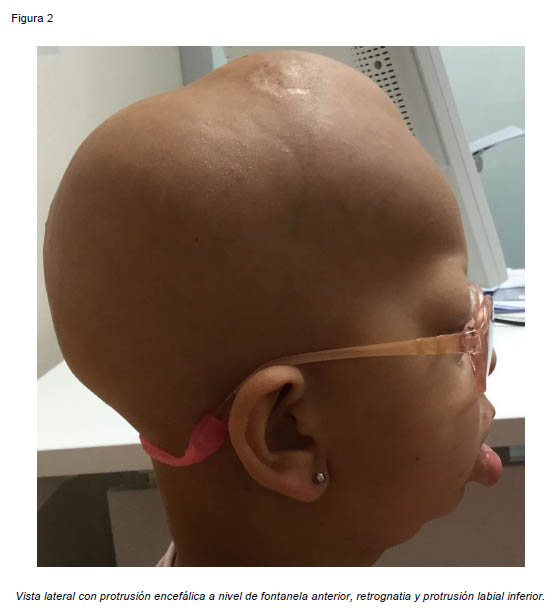
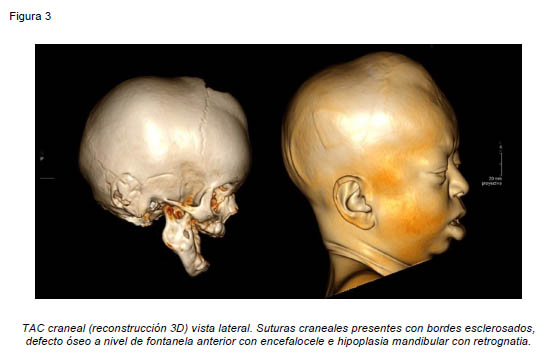
Presentamos el caso de una paciente de sexo femenino de 7 años de edad, derivada desde otro centro hospitalario para valoración de deformidad facial y retraso del crecimiento. En la exploración física se aprecia alopecia total, macroglosia, anodoncia, protrusión labial inferior, laxitud cutánea, talla baja, pectus Currarino-Silverman y braquidactilia en manos y pies. A nivel craneal presenta defecto óseo en fontanela anterior con protrusión encefálica, retrusión del tercio medio facial y barra supraobitaria originando exoftalmos e hipertelorismo, y retrognatia. La paciente ha sido intervenida en múltiples ocasiones por glaucoma bilateral y presenta actualmente nistagmus horizontal, fotofobia intensa, disminución de la agudeza visual, megalocornea y leucomas bilaterales (Figuras 1 y 2). Se realizó estudio radiológico inicial mediante Tomografía Axial Computarizada, objetivándose suturas craneales patentes con esclerosis de los bordes de forma generalizada y defecto de erupción dental (Figura 2). Por todo ello se estableció el diagnóstico de síndrome GAPO.
Discusión
GAPO es el acrónimo propuesto por Tipton y Gorlin en 1984, para designar el síndrome caracterizado por retraso del crecimiento, alopecia, pseudoanodoncia y atrofia óptica(2). Se trata de una rara enfermedad de la que se han descrito hasta el momento 35 casos aproximadamente. Se ha sugerido un patrón de herencia autosómica recesiva, y mutaciones del gen del receptor 1 de la toxina del ántrax
(ANTXR1) como origen etiológico. Este gen está ligado a la producción de una molécula que interviene en la homeostasis de la matriz extracelular(3), produciéndose un exceso de matriz extracelular del tejido conectivo que se acumula e interfiere con la función normal de los órganos. Se han descrito alteraciones a nivel cardiovascular, esquelético, cerebrovascular, pulmonar, auditivo y reproductivo(4), por lo que resulta fundamental su manejo multidisciplinar.
Bibliografía
- Salas-Alanís JC, Scott CA, Fajardo-Ramírez OR, Duran C, Moreno-Treviño MG, Kelsell DP. New ANTXR1 Gene Mutation for GAPO Syndrome: A Case Report. Mol Syndromol. 2016;7(3):160-3.
- Tipton RE, Gorlin RJ. Growth retardation, Alopecia, Pseudo-anodontia, and optic atrophy—The GAPO syndrome: Report of a patient and review of the literature. Am J Med Genet. 1984;19(2):209–216.
- Bayram Y, Pehlivan D, Karaca E, Gambin T, Jhangiani SN, Erdin S, et al. Whole Exome Sequencing Identifies Three Novel Mutations in ANTXR1 in Families with GAPO Syndrome. Am J Med Genet A. septiembre 2014;0(9):2328-34.
- Benetti-Pinto CL, Ferreira V, Andrade L, Yela DA, De Mello MP. GAPO syndrome: a new syndromic cause of premature ovarian insufficiency. Climacteric. noviembre 2016;19(6):594-8.
Figuras
|
