Introducción
Las alteraciones genéticas son causa frecuente de enfermedad, secuelas y muerte entre los lactantes y niños(1).
Se han identificado en el hombre tres clases de defectos genéticos.
- Mutación de un gen único (Monogénicas).
- Anomalías de los cromosomas (Cromosomopatias).
- Herencia multifactorial (Poligénicas).
El diagnóstico de un trastorno hereditario puede ser difícil cuando no existen otros parientes afectos por lo que el médico debe estar familiarizado con diferentes tipos de enfermedades genéticas y utilizar bibliografía para identificar patrones hereditarios.
Solo para las anomalías cromosómicas existen pruebas de laboratorio que proporcionan la evidencia del trastorno genético.
El conocimiento de que el número exacto de cromosomas en el hombre era de 46, en el año 1956 y el descubrimiento en 1959 de que el síndrome de Down se trataba de una trisomía del par 21, establecieron que las anomalías citogenéticas son un factor etiológico importante en la enfermedad humana(1).
En los 15 años siguientes se desarrollaron procedimientos de laboratorio que permitieron la precisa identificación de cada cromosoma individualmente y sus segmentos y facilitaron la interpretación tanto de las complejas fusiones cromosómicas como de las mínimas variaciones morfológicas.(1)
Todo este desarrollo ha creado una mayor demanda de análisis citogenético. Las indicaciones clínicas más importantes de estudio citogenético son las malformaciones congénitas, especialmente si afectan a más de un sistema y el retraso mental es de origen desconocido.
Se estima que 1 de cada 140 recién nacido presenta una anomalía cromosómica, además el hecho de que el 50–60% de los abortos espontáneos precoces se deban a anomalías cromosómicas sugiere que el menos el 7% de los embarazos humanos tienen una anomalía en el cariotipo y aproximadamente el 90% de las concepciones cariotípicamente anormales no llegan al término del embarazo(1),(2).
Las alteraciones cromosómicas pueden dividirse en dos tipos.(2),(3).
- Numéricas: son aquellas que determinan una alteración del complemento cromosómico normal, se dividen en dos grupos según afecten en su totalidad o parcialmente es decir por exceso o por defecto.
- Estructurales: se definen como cambios de la estructura normal del cromosoma, son heredables y cuando ocurren en los cromosomas de las células germinales se dividen en cinco tipos. Deleciones, Duplicaciones, Inversiones, Translocaciones e Isocromosomas.
Las deleciones son la pérdida de un fragmento del cromosoma, pueden ser terminales si es el fragmento distal el que se pierde, intersticial si es el fragmento intermedio entre el centrómero y el extremo germinal del cromosoma.
Entre las deleciones terminales tenemos la formación del cromosoma en anillo que se produce cuando ambos extremos del cromosoma se fragmentan y se unen.(3),(4)
Presentación del Caso
Se trata de un lactante de seis meses de edad, procedencia rural.
Antecedentes prenatales: embarazo normal.
Antecedentes natales: parto distócico. Cesárea por sufrimiento fetal agudo a las 38,5 semanas de gestación. Pesa al nacer 2400 gramos (CIUR) apgar 8-9.
Antecedentes post natales: síndrome de Distres Respiratorio al nacer que se mantiene junto al estridor laríngeo audible a distancia y en estado de reposo por lo que permanece ingresado en servicio de Neonatología hasta los 26 días que se envía al Hospital William Soler para descartar Anillo Vascular, diagnóstico que fue descartado regresando a la provincia, ingresa en sala de Terapia Intermedia y luego va a la sala de Respiratorio donde por la intensidad del estridor acompañado de dificultad respiratoria y cianosis ocasional se coordina con ORL y se realiza Laringoscopia exploratoria, así como otras investigaciones complementarias, al egreso por el riesgo se traslada para el Hospital del municipio de procedencia donde permanece hasta que logra mejor peso y alimentación sin dificultad ni riesgo.
Examen Físico
Aspecto de desnutrido, llanto agudo y débil.
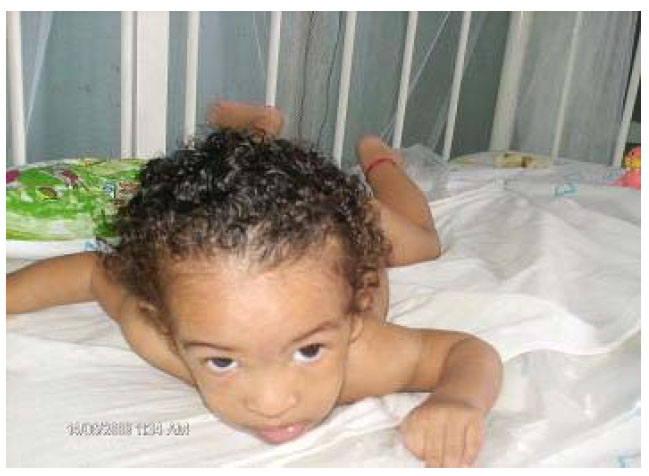
Facie: dismorfia craneofacial con asimetría facial. Hipoplasia de nares. Implantación baja del cuero cabelludo en la frente. Orejas bajas con rotación posterior. Micrognatia.
Respiratorio: estridor laríngeo audible a distancia, cianosis ocasional, disnea moderada con tiraje supra-esternal, supraclavicular y subcostal que se empeora cuando llora, ruidos trasmitidos y murmullo vesicular rudo. FR: 50’.
ACV: ruidos cardíacos rítmicos y enmascarados por el estridor.
Resto del Examen Físico: no aspecto de interés.
Exámenes complementarios
Hb: 112 g/ L Leucocitos 9.8 x 10 /L P 0,26 L 0.72 E 0.02
Coagulograma: normal.
Creatinina: 62 mmol /L
Urea: 1.4 mmol /L
Ácido Úrico: 20 mmol /L
TGP: 11UI
TGO: 12UI
Albúmina: 38 g/l
Proteínas totales: 49 g /L
Esofagograma: esófago con buena peristalsis, se descarta compresión extrínseca, llama la atención la constricción mantenida de la unión orofaríngea por debajo del cartílago cricoides.
Ecocardiograma: normal.
Rx de Tórax: normal.
Laringoscopia: se plantea Laringotraqueomalacia.
Cariotipo: 46, XY, del (5), (p15.1).
Discusión
El Síndrome del maullido de gato (Lejeune). Es consecutivo a una deleción de los brazos cortos de un cromosoma del par 5 (5p-)que incluye la región 5p14-15 que es la zona crítica para que se presente el síndrome (3),(4).
El Síndrome fue descrito por primera vez por Lejeune en 1963 y su frecuencia se calcula en 1/50 000 recién nacido, en publicaciones posteriores se han descrito más de 100 casos con esta cromosomopatía.(3).
En un principio se creyó que las deleciones cromosómicas resultaban letales para el ser humano, pero actualmente han sido documentados varios síndromes clínicos causados por ellos, algunos dan lugar a una afectación del fenotipo con menor severidad que las Trisomías.
Aproximadamente el 85% de los casos se debe a deleciones de novo esporádicas, en el 15% restante las deleciones son secundarias a la segregación desigual de una translocación parental. En el 85% de los casos en que el síndrome se debe a una deleción de novo, el cromosoma con deleción es de origen paterno. Aunque el tamaño de la deleción es variable.
Lo más característico es el llanto en la etapa de recién nacido y primeros meses que semeja al maullido de un gato, de ahí su nombre, son niños con escaso peso al nacer, hipotónicos, con retraso psicomotor, llanto especial y malformaciones diversas.
Signos clínicos más importantes de la deleción 5p-
Generales:
- Bajo Peso al nacer.
- Retraso mental.
- Llanto Agudo (maullido de gato) por hipoplasia laríngea.
- Crecimiento lento.
- Hipotonía.
Craneofaciales:
- Microcefalia con asimetría craneal.
- Cara redonda de luna llena.
- Manifestaciones oculares: hipertelorismo, epicantus, hendidura palpebral antimongoloide, estrabismo, miopía y atrofia óptica.
- Hipoplasia de los huesos nasales con nariz ancha y plana.
- Orejas grandes de implantación baja, con tubérculos pre-auriculares.
- Micrognatia.
Tórax:
- Cardiopatía congénita ocasional
Pelvis y Abdomen:
- Hernia inguinal.
- Diástasis de rectos.
- Palas ilíacas pequeñas.
- Luxación de caderas.
Extremidades:
- Metacarpianos y Metatarsianos pequeños.
- Sindactilia parcial.
- Clinodactilia.
- Pie plano o zambo.
- Surco simiesco.
- Laxitud ligamentosa.
Otros Signos
Pueden presentarse malformaciones digestivas, cardiovasculares, renales, genitales, cerebrales y vertebrales. Es casi constante la presencia de Hipoplasia de laringe responsable del llanto peculiar de estos niños.
En los dermatoglifos es frecuente la presencia de pliegue palmar único, trirradius palmar en posición t y ausencia o verticalidad del trirradius del cuarto dedo.
Cuanto mayor sea la deleción menor será la inteligencia, la estatura y el peso. Se ha calculado que aproximadamente el 1% de los pacientes institucionalizado con retraso mental lo padecen.
El pronóstico en cuanto a la vida es bueno, dependiendo lógicamente de la gravedad de las malformaciones asociadas y de la dificultad respiratoria y digestiva.
Las malformaciones clínicas y el grito característico se modifican con la edad, acentuándose el retraso psicomotor, la cara se alarga, el epicantus se atenúa y aparece hipertonía y espasticidad.
Prevención y terapéutica
Es fundamental el consejo genético. Si hay translocaciones en uno de los progenitores, la cuarta parte de los hermanos estarán afectados, además se debe estimular el desarrollo para mejorar el rendimiento psíquico.
Anexos


Referencias
- Hall J G. Anomalías cromosómicas clínicas. En: Behrman RE. Nelson Tratado de Pediatría. 17 ed. Madrid: Elsevier; 2005. p. 382 – 390.
- Ballesta F., Cruz M. Cromosomopatías. En: Cruz Hernández M. Tratado de Pediatría. 8 ed. Madrid: Ediciones Ergom; 2001. p. 253 – 263.
- Thompson J S, Thompson MW. Aberraciones cromosómicas. En: Genética Médica. 3 ed. Ciudad de la Habana: Editorial Científico Técnico; 1986. p. 164 – 178.
- Smith. Patrones reconocibles de malformaciones humanas. p. 40 – 43.
- Leon López R. et al. Síndrome del Maullido del Gato. Presentación de un caso. Rev. Cub. MGI, oct – sept; 1995.
- Goodman – Garlin H. Síndromes Genéticos: The Cri – Du - Chat. Syndrome. Hom Gent 1978; 42: p. 143 – 152.
- Breg W, Steele M. Miller O. The Cri – Du – Chat Syndrome in adolescents and patient wiht partial delection of the short arm of chromosome 5 (5p-). J Pediatr 1970; 77: p. 782 – 91.
|
