Introducción
Los trastornos neuromusculares son motivo de consulta a diferentes especialistas infantiles. Los pediatras son los primeros en ser consultados, y es frecuente la referencia a ortopedistas infantiles y otros especialistas que acogen las muy variadas consultas cuyo estudio conduce finalmente al diagnóstico de un trastorno neuromuscular.
Estas enfermedades no son infrecuentes dentro de la morbilidad pediátrica, presentando incidencias de: 1 en 3.500 recién nacidos vivos hombres en distrofia muscular de Duchenne (DMD) y de 1 en 6.000 nacidos vivos en atrofias musculares espinales (AME) (1). Se encuentran ampliamente distribuidas a nivel mundial, especialmente en zonas de alta consanguinidad. Muchas de ellas son causa de discapacidad progresiva en el niño, lo que genera alto impacto a nivel individual, de la familia y la sociedad, por lo cual un diagnóstico oportuno es fundamental. Sin embargo, la multiplicidad de formas de presentación y la poca especificidad de los síntomas iniciales determina tardanza en el diagnóstico (2). El médico pediatra general o de especialidades relacionadas juega un rol relevante en este aspecto, ya que son los responsables de reconocer precozmente estas enfermedades y de iniciar las acciones necesarias para un diagnóstico y manejo terapéutico oportunos.
¿Qué son los trastornos neuromusculares y cómo clasificarlos?
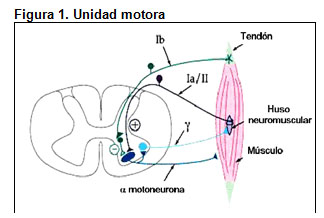
Los trastornos neuromusculares constituyen un grupo de enfermedades que afectan cualquiera de los componentes de la unidad motora, es decir, la unidad funcional constituida por el cuerpo de la motoneurona del asta anterior de la médula espinal, su axón (nervio periférico) y todas las fibras musculares inervadas por esta motoneurona (Figura 1).
En algunos cuadros, además del compromiso de la unidad motora existe compromiso de otros tejidos (cerebro, corazón, etc.) por lo que muchos de estos trastornos son considerados actualmente “multisistémicos” (3).
El efector final es el músculo, pero éste puede comprometerse en forma primaria, o secundaria a la denervación. De acuerdo a este concepto las ENM pueden clasificarse en:
1. Atrofias neurogénicas que incluyen las enfermedades de la motoneurona y del nervio (neuropatías).
2. Miopatías o patologías primarias del músculo sin alteraciones estructurales en nervio periférico.
3. Trastornos de la unión neuromuscular.
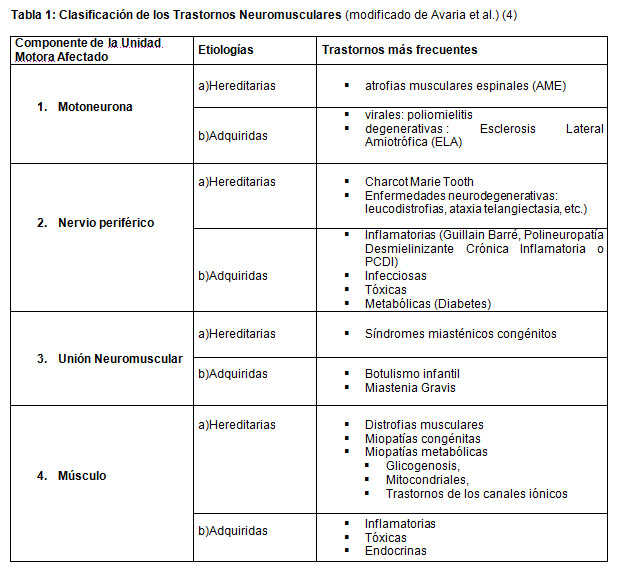
Cada una de estas afecciones puede ser de causa hereditaria o adquirida, dando origen a una clasificación que se presenta en la tabla 1.
¿Cómo se presentan los trastornos neuromusculares?
Las formas de presentación (Tabla 2) dependen de la edad de inicio y de las características de cada enfermedad, y según su curso pueden ser agudas o crónicas.
De esta amplia variedad de formas de presentación, la mayoría inespecíficas, surge el enfrentamiento al paciente con un probable trastorno neuromuscular, que incluye varias etapas:
1. Definir la anormalidad (¿cuál es el problema?): Para ello debemos definir el (los) motivo(s) de consulta (síntomas) y el conjunto de signos al examen neurológico que permiten establecer un diagnóstico sindromático (miopático, neuropático, etc.).
2. Definir la localización (¿dónde está el problema?): establecer el diagnóstico topográfico (motoneurona, axón, mielina, diferentes elementos de la unión neuromuscular, músculo).
3. Definir la etiología (¿cuál es la causa del problema?):fundamentalmente dirigido a diferenciar cuadros genéticamente determinados o adquiridos. En el niño los cuadros genéticamente determinados son mucho más frecuentes que los adquiridos, especialmente en el recién nacido, sin embargo es imprescindible considerar las causas adquiridas, dado que en su mayoría tienen tratamiento curativo y mucho mejor pronóstico.
4. Revisar el diagnóstico diferencial (¿qué otros cuadros podemos descartar?): Esta etapa del abordaje al paciente neuromuscular es esencial por cuanto las ENM de distinta etiología pueden compartir historia, semiología como también algunos hallazgos de laboratorio, lo que obliga a, que pese a que el diagnóstico parezca obvio, plantear diagnósticos diferenciales y argumentos por los cuales se descartan otras posibilidades.
No siempre es posible definir simultáneamente diagnósticos sindromáticos, topográficos y etiológicos, sin embargo el establecer síndrome, localización y diagnóstico diferencial nos permite trazar un plan de estudio y tratamiento racional.
¿Qué investigamos en la anamnesis?
1. Identificamos el motivo de consulta principal (debilidad, retraso motor, etc.) intentando establecer si traduce una alteración de la unidad motora.
2. Interrogamos acerca de motivos de consulta asociados que, pueden o no, estar relacionados con el motivo de consulta principal. Por ejemplo en un paciente con una distrofia muscular de Duchenne (DMD), el motivo de consulta puede ser el retardo motor (niño hombre que no camina a los 18 meses), trastorno de la marcha (marcha bamboleante con caídas frecuentes) o la debilidad proximal (dificultad para pararse del suelo o para subir escaleras); el motivo de consulta asociado (que a veces puede ser el más importante para los padres) puede ser un retraso de lenguaje o dificultades aprendizaje evidenciadas precozmente en la etapa preescolar y de ocurrencia frecuentemente en esta distrofia muscular. En general estos niños presentan coeficiente intelectual menor que el promedio de la población general y en cerca de un tercio se evidencia un retardo mental leve. Los motivos de consulta asociados o “secundarios” son claves en el diagnóstico y no deben omitirse o atribuirse a retrasos madurativos. Este tipo de errores en la interpretación de los síntomas secundarios retrasará el diagnóstico, frecuentemente más allá de los 4 o 5 años, lo que en muchas familias implica un consejo genético tardío.
3. Definimos la edad de inicio de los síntomas, que permite orientarse hacia grupos de causas. La edad de adquisición de distintos hitos del desarrollo psicomotor (DSM) nos orienta a este respecto y acerca de etiología y evolución del cuadro. En DMD 50% de los niños camina tardíamente más allá de los 18 meses. En Atrofia Muscular Espinal los hitos del DSM adquiridos son esenciales para diferenciar entre los distintos tipos de AME: AME tipo 1 pueden o no adquirir control cefálico, pero no logran sedestación autónoma; AME tipo 2 logran sedestación, pero no marcha autónoma; AME tipo 3 logran marcha autónoma (5).
a) La debilidad de inicio desde el nacimiento (manifestada como hipotonía, hipomovilidad, dificultades respiratorias o de deglución, apuntará a Distrofia Miotónica Congénita, miopatías congénitas, distrofias musculares congénitas y menos frecuentemente a una neuropatía congénita por hipomielinización o algunas formas de enfermedades de motoneurona. Aún menos frecuentes, los síndromes miasténicos congénitos deben considerarse ya que pueden mejorar con anticolinesterásicos.
b) La debilidad de inicio en el preescolar orienta a algunas distrofias musculares, atrofias musculares espinales tipo 2 o 3 o algunos cuadros adquiridos, como las miopatías o polineuropatías inflamatorias. En el escolar son más frecuentes las polineuropatías.
4. Establecemos el perfil temporal del cuadro, que orientará hacia la etiología. Se consideran perfiles temporales agudos aquellos que alcanzan la máxima debilidad en días o semanas (menos de 1 mes en el caso de las neuropatías inflamatorias), subagudos en pocas semanas a meses, y crónicos que evolucionan en años. En general cuadros de perfil temporal agudos o subagudos obligan a plantear en primer término causas adquiridas como inflamatorias, tóxicas o endocrinas. Excepciones a esto son algunas de las miopatías metabólicas genéticamente determinadas que pueden debutar en forma aguda con debilidad o intolerancia al ejercicio.
5. Igualmente importante es definir si la evolución de los síntomas es estacionaria (síntomas estables durante largos periodos de tiempo), progresiva (como ocurre en la gran mayoría de las distrofias musculares progresivas) o fluctuante (característico del compromiso de la unión neuromuscular, miopatías metabólicas y canalopatías).
6. Otros aspectos a investigar son antecedentes del embarazo, patología obstétrica, percepción de movimientos fetales tardíos o disminuidos, distocia de posición, polihidroamnios, antecedentes de abortos repetidos o muerte neonatal precoz. Cualquiera de estos antecedentes puede sugerir morbilidad neuromuscular. Antecedentes del período neonatal como hipotonía dificultades respiratorias o deglutorias, aun leves o transitorias, pueden ser orientar a un trastorno neuromuscular en especial la Distrofia Miotónica, que en su forma congénita puede ser oligosintomática.
7. En cuadros de debilidad de inicio agudo o subagudo en el escolar o adolescente es necesario investigar dirigidamente exposición a tóxicos (alcohol, plomo, arsénico, N-hexano) o fármacos que pueden producir una polineuropatía sensitivo motora adquirida.
8. Del mismo modo es necesario evaluar enfermedades asociadas:
a) Diabetes: es causa de neuropatía en cuadros de algunos años de evolución y se asocia a cuadros neuromusculares con compromiso multisistémico como la Distrofia Miotónica de Steinert y las citopatías mitocondriales
b) Hipoacusia: se asocia a varias enfermedades neuromusculares, entre las que se incluyen algunas neuropatías hereditarias, miopatías mitocondriales, enfermedad de Refsum, etc.
c) Cardiopatía: de relativa frecuencia en distrofinopatías, distrofias facio-escápulo-humeral, distrofia de Emery Dreifuss, laminopatías, miopatías metabólicas, mitocondriales y otras.
d) Compromiso ocular: de cámaras (distrofias musculares congénitas con compromiso ocular), cataratas (asociadas a neuropatías específicas y distrofia miotónica entre otras), etc.
Es importante tener presente que estas alteraciones asociadas pueden ser parte de trastornos multisistémicos familiares en que los síntomas pueden presentarse en diferentes momentos en individuos de una misma familia, de modo que deben buscarse en forma dirigida y reiterada en el paciente y su familia.
9. Finalmente es de particular importancia la historia familiar: se debe recoger información detallada y realizar árbol genealógico completo, consignando individuos afectados y no afectados, patología central y otras aparentemente no relacionadas, poniendo especial énfasis en sexo de los afectados, severidad de los síntomas en generaciones sucesivas y consanguinidad. Con varios individuos afectados, es posible definir el tipo de herencia: autosómica dominante o recesiva, ligada al X, o mitocondrial. Los casos aislados pueden corresponder a trastornos recesivos o ligados al X con bajo números de hijos en distintas generaciones (familia no informativa). La ausencia de antecedentes familiares no descarta una enfermedad hereditaria, pudiendo corresponder a mutaciones nuevas de cuadros con alta tasa de mutaciones (distrofias distrofinopáticas), a casos de paternidad dudosa, o casos en que no hay información completa de la familia disponible. Es muy importante tener presente que la información relativa a los familiares afectados puede no surgir espontáneamente o incluso puede ser negada en el caso de enfermedades hereditarias cuyas implicancias diagnósticas y pronósticas pueden constituir una carga emocional significativa para el o los familiares afectados. En estos casos la obtención de la historia familiar puede requerir todo un proceso de investigación cuidadoso y progresivo.
¿Qué buscamos en el examen físico?
1. El examen se inicia con la inspección con énfasis en:
a) Aspecto facial y general: La debilidad facial es frecuente en las Distrofia Miotónica y Facio-escápulo-humeral y la miopatía nemalínica. Habitualmente confiere un aspecto especial con facies alargada, escasa mímica facial, boca entreabierta o en “V” invertida. Por el contrario cuadros de motoneurona que cursan con severa debilidad de extremidades como la Atrofia Muscular Espinal tipo 1 (Werdnig Hoffman) tienen característicamente indemnidad facial con excelente fuerza y mímica facial.
b) Trofismo: observamos la constitución y masa muscular en general, buscando hipo o hipertrofias, precisando su distribución y asimetrías que puedan dar cuenta de atrofias monomiélicas (de una extremidad) o localizada en segmentos proximales, distales o de algunos segmentos específicos como la lengua, cuya atrofia sugiere denervación por compromiso de nervios hipoglosos o de las motoneuronas del núcleo de este par bulbar. Algunas distrofias musculares presentan patrones particulares de atrofia como la atrofia selectiva del compartimiento posterior del muslo en las calpainopatías o atrofia del compartimiento posterior de la pierna en las disferlinopatías. La hipertrofia muscular es también un signo característico de ciertos cuadros musculares: hipertrofia generalizada que confiere a los niños un aspecto “musculoso” puede observarse en canalopatías como la Miotonía Congénita de Thomsen o localizada en gemelos, vasto lateral de cuádriceps, lengua, deltoides y glúteos en la Distrofia de Duchenne. Estos signos son muy importantes dado que pueden estar presentes antes que la debilidad o los problemas motores sean significativos.
c) Alteraciones ortopédicas: El pie cavo, bot, plano y la escoliosis son frecuentes en cuadros neuromusculares. El pie cavo se observa en las polineuropatías hereditarias y si es asimétrico obliga a buscar una disrafia. Pie bot es la contractura aislada más frecuente y puede estar presente en disrafias, distintos tipos de artrogriposis, distrofias musculares congénitas y es un signo clave en la Distrofia Miotónica Congénita oligosintomática.
d) Fasciculaciones, descritas como movimiento “en bolsa de gusanos”, son signo de denervación. Su presencia en la lengua traduce denervación por compromiso de nervios hipoglosos. Temblor postural y/o de acción puede observarse en algunas polineuropatías hereditarias. El poliminimioclonus es un fino temblor distal de los dedos, frecuente en atrofias espinales, secundario a fasciculaciones de los músculos intrínsicos de la mano.
e) Postura y marcha: La marcha anadina (de pato) o con bamboleo de la pelvis refleja debilidad de músculos proximales (abductores, extensores de cadera y cuádriceps) y orienta a miopatías. La marcha estepada (steppage en inglés)se caracteriza por la elevación y flexión exagerada del muslo sobre la pelvis para levantar la extremidad distal y “lanzarla” hacia delante a causa de la debilidad de los músculos distales como el tibial anterior y perineos. Esta marcha es característica de las polineuropatías y de algunas miopatías distales menos frecuentes.
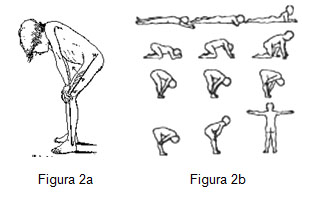
f) Finalmente en la inspección observamos la forma como se incorpora el niño del suelo. Niños con debilidad proximal emplean para hacerlo la maniobra de Gowers, en que se levantan apoyando las manos sobre los muslos, como si treparan sobre sí mismos (figura 2a y 2b).
g) El examen debe incluir marcha, en puntas de pie y en talones, carreras y la incorporación desde el suelo, ya que grados menores de debilidad pueden pasar desapercibidos sin estas maniobras.
2. El examen continúa con la evaluación de fuerza muscular en que se evalúan músculos proximales y distales de extremidades, considerando inervación y definiendo si la distribución de la debilidad apoya compromiso de motoneurona, de raíz, plexo, nervio periférico, unión neuromuscular (UNM) o músculo. Los grupos musculares habitualmente examinados en extremidades superiores incluyen deltoides, bíceps, tríceps, flexores y extensores de muñeca. En extremidades inferiores se evalúan glúteos mayores (extensores de cadera), glúteos medios (abductores de cadera), cuádriceps, tibiales anteriores, gemelos y peroneos. Si sospechamos compromiso selectivo de un nervio (mononeuropatía), de plexo (plexopatía) o raíz (radiculopatía) haremos el examen dirigido a los músculos inervados por el nervio, raíz o plexo en cuestión.
La distribución es especialmente importante para establecer la localización del compromiso en la unidad motora, y si bien existen excepciones, las miopatías producen en general debilidad de predominio proximal en tanto que las polineuropatías debilidad de predominio distal.
Para cuantificar fuerza, la escala más utilizada y probablemente con menor variación inter.-examinador es la Escala del Medical ResearchCouncil que aparece en la tabla 3.
3. La palpación del músculo permite definir consistencia, sensibilidad, calcificaciones y contracturas. En muchas distrofias musculares los músculos se palpan de consistenciagomosa (“como caucho”) en las zonas de hipertrofia muscular. Miopatías metabólicas o inflamatorias pueden presentar sensibilidad dolorosa a la palpación de los músculos afectados.
4. El examen de movilidad articular permite identificar contracturas que aparecen en algunas articulaciones producto del desbalance muscular originado por músculos antagonistas que se comprometen en grado distinto. Las contracturas son más frecuentes en las distrofias musculares y poco prominentes o tardías en las enfermedades de motoneurona. En distrofia de Duchenne las contracturas más precoces son las de flexores de cadera (desbalance entre flexores y extensores de cadera) y de tobillos (desbalance de dorsiflexores y flexores plantares del pie).
5. La percusión del músculo permite evidenciar miotonía o dificultad del músculo para relajarse, la que puede ser evocada por la acción del músculo (por ejemplo empuñar las manos para observar la dificultad en la posterior extensión de los dedos) o por la percusión directa del músculo con el martillo de reflejos. Esto puede buscarse en músculos de extremidades o en la lengua. La miotonía es un fenómeno característico en las miotonías congénitas, algunas de las Parálisis Periódicas y en la Distrofia Miotónica de Steinert en la que puede no estar presente en el recién nacido, por lo que debe buscarse en los familiares (especialmente la madre).
6. El examen de tono permite establecer la presencia de hipotonía, característica de la mayoría de los cuadros neuromusculares y que en los trastornos de la unidad motora es proporcional al grado de debilidad.
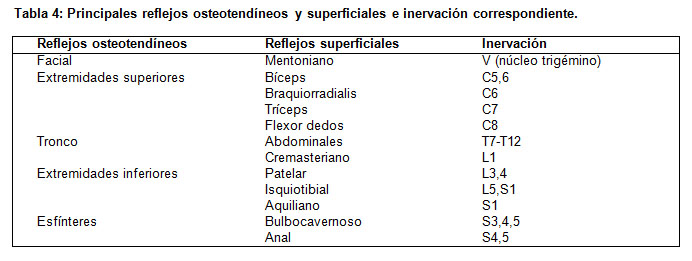
7. El examen de los reflejos osteotendíneos (ROT) permite definir la topografía de la afección. En las enfermedades de motoneurona la hipo o arreflexia es global o de predominio proximal, en las neuropatías es de predominio distal y en las miopatías los reflejos distales se encuentran más vivos que los proximales. En el caso de las radiculopatías o plexopatías la hipo o arreflexia dependerá del segmento comprometido. Los principales reflejos osteotendíneos (ROT) y su nivel de integración correspondiente se resumen en la tabla 4.
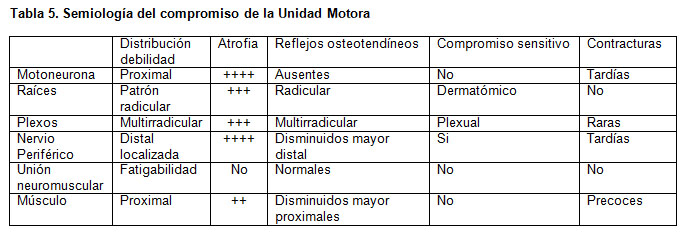
Los signos del examen más característicos de compromiso de los distintos componentes de la unidad motora se resumen en la tabla 5.
¿Qué estudios de laboratorio tenemos disponibles para el estudio de los trastornos neuromusculares?
Los exámenes de utilidad en el estudio de las ENM tienen indicaciones y utilidad precisas. Su rendimiento se relaciona directamente con una buena indicación, interpretación y conocimiento de sus limitaciones.
- Enzimas Musculares: Creatinkinasa o creatin-fosfoquinasa (CPK)
- Estudios electro-fisiológicos
- Velocidad de conducción nerviosa
- Electromiografía
- Estudios (electromiografía) de fibra única
- Test estimulación repetitiva
- Estudios de respuesta a anticolinesterásicos:
- Test de Tensilón MR (cloruro de edrofonio) o neostigmina
- Estudios histopatológicos
- Biopsia muscular
- Biopsia de nervio
- Estudios imagenológicos
- Ecografía, tomografía axial computada (TAC) y resonancia magnética de músculos
- Estudios bioquímicos y genéticos específicos.
Enzimas musculares
Varias enzimas son útiles en el estudio de las enfermedades musculares: transaminasas, lactato dehidrogenasa (LDH), aldolasa y creatinkinasa o creatinfosfokinasa (CK). La CK existe en tres isoformas: la isoforma BB existe sólo en encéfalo, la MM en tejido muscular esquelético y cardíaco y la fracción MB se encuentra en varios tejidos. El músculo esquelético, aunque contiene preferentemente la fracción MM, contiene además alrededor de un 5% de isoenzima MB lo que aumenta hasta un 25% en el músculo inmaduro normal, por lo que sus resultados deben ser cuidadosamente interpretados en el recién nacido (7). Los eritrocitos y el hígado no contienen esta enzima, por lo que la hemólisis y la disfunción hepática no aumentan los valores de CK. La necrosis de fibras musculares, característica de las distrofias musculares, aumenta la concentración sérica de CK, pero es necesario considerar que también lo aumentan el trauma muscular y el parto (vaginal o cesárea) especialmente durante las primeras 30 horas post parto (8).
Imagenología
Ecografía, TAC y resonancia magnética de músculo se han utilizado en la localización de músculo anormal para dirigir la biopsia, teniendo especial utilidad en el diagnóstico de miopatías inflamatorias. Las imágenes de músculo han cobrado importancia ya que permiten identificar el compromiso selectivo de algunos grupos musculares en los diferentes segmentos y orientar el estudio genético molecular.
Estudio electrofisiológico
El estudio electrofisiológico es de utilidad para orientar el diagnóstico en casos clínicamente poco definidos de síndrome hipotónico. Sin embargo, hay que considerar que los resultados de estos análisis no son siempre específicos, dependen de la experiencia del examinador y por sobre todo deben ser interpretados de acuerdo a la clínica del paciente.
La evaluación electrofisiológica incluye el estudio de conducción nerviosa sensitiva y motora y el registro de la actividad eléctrica muscular mediante electrodos de aguja introducidos en el músculo (9).
En la electromiografía se analiza:
1. Actividad de inserción, que corresponde al daño mecánico provocado en las fibras con la inserción de la aguja, generalmente aumentada en procesos inflamatorios.
2. Presencia de actividad espontánea en reposo, lo que es anormal, especialmente la presencia de fasciculaciones, fibrilaciones y ondas positivas, secundarias a denervación, especialmente por compromiso de motoneurona inferior. La inserción de la aguja puede provocar descargas repetitivas de potenciales de acción como en las miotonías, o ráfagas de potenciales de denervación (ondas positivas).
3. Potenciales de acción de unidad motora. La activación del músculo provoca descarga de potenciales de acción de unidad motora (PAUM) voluntarios que son analizados en cuanto a su duración, amplitud y fases. Potenciales de acción muscular breves, pequeños y polifásicos son característicos de los procesos miopáticos, pero se encuentran con mayor frecuencia en el niño que en el adulto normal y deben interpretarse con precaución, de acuerdo a su frecuencia y distribución. En los procesos neuropáticos crónicos es común encontrar potenciales de mayor amplitud, anchos y polifásicos.
Patrón de interferencia. Con el aumento de esfuerzo muscular se produce reclutamiento proporcional y ordenado de unidades motoras, generando un patrón de interferencia completo cuando las unidades motoras individuales no pueden reconocerse. Un patrón de interferencia completo con esfuerzo moderado o leve es característico de los procesos miopáticos primarios, en que una gran cantidad de unidades motoras deben reclutarse para generar este esfuerzo muscular mínimo. Por el contrario, en los procesos neuropáticos se registra un patrón de interferencia simple, en que pocas unidades motoras descargan a frecuencia aumentada. (10)
El estudio de velocidad de conducción nerviosa (VCN) se basa en la obtención de potenciales de acción muscular mediante la estimulación eléctrica de un nervio en dos puntos distintos y midiendo el tiempo de aparición de ambos (latencias). Dividir la distancia recorrida por este valor (tiempo) permite obtener la velocidad de conducción del impulso nervioso. Los valores de velocidad de conducción nerviosa dependen de la edad, siendo en el recién nacido del 50% de lo observado en el adulto y alcanzando los valores del adulto cerca de los 4 años. (11)
Las anormalidades de la conducción nerviosa permiten dividir las polineuropatías en desmielinizantes,que presentan prolongación de las latencias y disminución de las velocidades de conducción nerviosa; y axonales, en que las latencias y las velocidades son normales pero las amplitudes de los potenciales están significativamente reducidas.
Histopatología
La biopsia muscular puede obtenerse mediante punción o una intervención quirúrgica a cielo abierto. La muestra debe ser manejada adecuadamente, en fresco, congelada en isopentano enfriado en nitrógeno líquido y sometida a tinciones corrientes y técnicas de histoquímica. Si se sospecha una miopatía congénita el examen de elección es la biopsia muscular con técnicas de histoquímica que permiten diferenciar tipos de fibras y anormalidades estructurales, (cuerpos de nemalina, núcleos centrales) y procedimientos que permitan el análisis de proteínas estructurales de la membrana de la fibra muscular como la distrofina en distrofias distrofinopáticas (Duchenne y Becker), merosina en la Distrofia Muscular congénita, y otras proteínas asociadas (sarcoglicanos, distroglicanos, disferlina) que se relacionan a diferentes tipos de distrofias de cinturas.
Genética molecular
El diagnóstico definitivo de muchas de las ENM se realiza actualmente mediante análisis de ADN, como en Atrofia Muscular Espinal y Distrofia Miotónica. Es importante recordar que los estudios genéticos tienen limitaciones, puesto que no permiten distinguir entre fenotipos leves y severos causados por la misma alteración genética y en esos casos el pronóstico sigue dado por el análisis del conjunto de elementos clínicos y de laboratorio. Ejemplo de esta situación es el estudio genético en AME, que si bien posibilita el diagnóstico de certeza de la AME asociada al cromosoma 5, no permite diferenciar AME tipo 1, 2 o 3. Lo mismo ocurre en el análisis de deleciones en el gen de la distrofina que tiene una sensibilidad limitada (positividad que no supera el 60%) y no permite diferenciar entre Duchenne y Becker. (12)
Otros estudios:
Una larga lista de exámenes tendientes a pesquisar las complicaciones que se asocian a los diferentes trastornos neuromusculares es parte del estudio habitual. Exploraciones respiratorias, nutricionales, endocrinas, gastrointestinales, cardíacas, ortopédicas y otras van a depender de la patología y de las complicaciones más frecuentemente relacionadas a ella. Ejemplos clásicos son: estudio cognitivo (en busca de compromiso intelectual característico), cardiaco y respiratorio en Distrofia de Duchenne; estudio auditivo, oftalmológico y cardíaco en distrofia facio-escápulo-humeral y estudio oftalmológico, endocrino y cardiaco (buscando trastornos del ritmo) en Distrofia Miotónica.
Síntesis
- Las herramientas más importantes en el estudio de las ENM en Pediatría son la anamnesis y el examen clínico neuromuscular, completo y detallado. Este abordaje eminentemente clínico permite:
- establecer diagnóstico sindromático
- orientar hacia cuadros específicos más probables
- dirigir el estudio a solicitar
- El laboratorio neuromuscular ayuda al diagnóstico diferencial entre fenotipos comunes y, si bien ha contribuido al mejor conocimiento e identificación de estas patologías, es esencial conocer sus indicaciones, aportes y limitaciones.
- El esfuerzo diagnóstico tiene como objetivos establecer un consejo genético, definir el pronóstico aproximado según las posibilidades terapéuticas disponibles a nivel mundial y establecer un plan terapéutico actualizado y orientado a mantener función, prevenir complicaciones y mejorar calidad de vida.
- Tan importante como lo anterior es identificar cuadros de menor prevalencia que los genéticamente determinados en los niños, pero la mayoría con tratamiento efectivos.
Referencias
- A. Population frequencies of inherited neuromuscular diseases – a world survey. Neuromuscular Disorders 1991; 1: 19-29.
- Avaria M.A., Kleinsteuber K., Herrera L., Carvallo P. Tardanza en el diagnóstico de la distrofia muscular de Duchenne en Chile. Rev Med Chile 1999; 127: 65-70.
- Goebel H.H. Advances in Understanding Hereditary Neuromuscular Diseases. Semin Pediatr Neurol 2002; 9 (2): 100-159.
- Avaria M.A, Kleinsteuber K., Castiglioni C. Enfermedades Neuromusculares en el recién nacido en Menéndez P., Hernández M., Pinto F. Neurología Perinatal, Editorial Medigraphia 2002, Capítulo 21, páginas 274-305.
- Munsat T., Davies K. Meeting report: International SMA Consortium meeting. Neuromusc. Disord. 1992; 2: 423 - 428.
- Brooke M. Motor unit weakness. En: Bradley W., Daroff R., Fenichel G., Mardsen C. eds. Neurology in Clinical Practice. Butterworth-Heinemann, Boston, 1991; 2: 275-288.
- Bodenstain J., Zellweger H. Creatinphosphokinase in normal neonates and young infants. J.Lab. Clin. Med.1971; 77: 853.
- Jedeikin R., Makela S., Shennan A., Rowe R., Ellis G. Creatine kinase isoenzymes in serum from cord blood and the blood of healthy full-term infants during the first three postnatal days. Clin Chem 1982 28(2): 317-22.
- Electrophysiologic testing and laboratory aids in the diagnosis of neuromuscular diseases. In Principles of Neurology Raymond Adams, Maurice Victor, Allan Ropper (eds) Chapter 45: 1285 Sixth edition McGraw Hill 1997.
- Royden-Jones H. Electromyographic evaluation of the floppy infant. In: Pediatric clinical electromyography. Royden-Jones H, Bolton C. Harper CM (Eds) Lippincott –Raven Publishers, Philadelphia, 1996.
- Cruz M., Perez C., Del Campo F., Barrio M., Gutierrez A., Lopez E. Sensory and mixed conduction velocity in infancy and childhood. I normal parameters in median ulnar and sural nerves. Electromyogr Clin Neurophysiol 1978; 18: 487-504.
- Miller R., Hoffman E. Molecular diagnosis and modern management of Duchenne muscular dystrophy. Neurol Clin 1994; 12(4), 699-725.
|